The 'cran_repo' argument in shelf() was not set, so it will use
cran_repo = 'https://cran.r-project.org' by default.
To avoid this message, set the 'cran_repo' argument to a CRAN
mirror URL (see https://cran.r-project.org/mirrors.html) or set
'quiet = TRUE'.
Warning: package 'reticulate' was built under R version 4.3.3
Warning: package 'bio3d' was built under R version 4.3.3
# source files and convert to notebooks for display in articlelapply(list('R/theme.R', 'R/rst.error.R', 'R/dssr.R','R/chiR.R', 'R/nuR.R', 'R/Contact_analyzer.R'),function(x) {source(x)r_to_qmd(x, paste0('notebooks/', gsub('R/', '', x), '.qmd')) })
# read all final minimization pdb filesmin.pdb.list <-lapply(list.files(path ='data', pattern ="5_min_dop.*\\.pdb$", full.names =TRUE), read.pdb)# concatenate xyz coordinatesfor (i in1:length(min.pdb.list)) {if (i ==1) { xyz <- min.pdb.list[[i]]$xyz[1,] } else { xyz <-rbind( xyz, min.pdb.list[[i]]$xyz[1,] ) }}
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Warning in rbind(xyz, min.pdb.list[[i]]$xyz[1, ]): number of columns of result
is not a multiple of vector length (arg 2)
Retaining 938 non-water atoms
Removing a total of 27966 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt02.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 37252"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 37252
.. number of calphas in PDB: 0
.. number of residues in PDB: 9152
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 36636 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt03.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 36364"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 36364
.. number of calphas in PDB: 0
.. number of residues in PDB: 8930
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 35748 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt04.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 34168"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 34168
.. number of calphas in PDB: 0
.. number of residues in PDB: 8378
Retaining 932 non-water atoms
Retaining 612 non-hydrogen atoms
Removing a total of 33556 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt05.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 36656"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 36656
.. number of calphas in PDB: 0
.. number of residues in PDB: 9003
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 36040 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt06.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 36972"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 36972
.. number of calphas in PDB: 0
.. number of residues in PDB: 9082
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 36356 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt07.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38038"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38038
.. number of calphas in PDB: 0
.. number of residues in PDB: 9350
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37420 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt08.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 35250"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 35250
.. number of calphas in PDB: 0
.. number of residues in PDB: 8650
Retaining 934 non-water atoms
Retaining 614 non-hydrogen atoms
Removing a total of 34636 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt09.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 37192"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 37192
.. number of calphas in PDB: 0
.. number of residues in PDB: 9137
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 36576 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt10.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 37962"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 37962
.. number of calphas in PDB: 0
.. number of residues in PDB: 9331
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37344 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt11.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 36728"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 36728
.. number of calphas in PDB: 0
.. number of residues in PDB: 9021
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 36112 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt12.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38566"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38566
.. number of calphas in PDB: 0
.. number of residues in PDB: 9482
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37948 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt13.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 36432"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 36432
.. number of calphas in PDB: 0
.. number of residues in PDB: 8947
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 35816 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt14.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 33620"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 33620
.. number of calphas in PDB: 0
.. number of residues in PDB: 8241
Retaining 932 non-water atoms
Retaining 612 non-hydrogen atoms
Removing a total of 33008 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt15.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 35920"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 35920
.. number of calphas in PDB: 0
.. number of residues in PDB: 8819
Retaining 936 non-water atoms
Retaining 616 non-hydrogen atoms
Removing a total of 35304 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
# create a xyz.list that contains all xyz xyz.list <-lapply(1:15, function(i) { pdb.traj <-get(paste0('pdb.traj.', sprintf('%02d', i)))return(pdb.traj$xyz)})
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 2246"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart5.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 3847"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 2246"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doprstslt01_200_restart5.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 3847"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Retaining 618 non-hydrogen atoms
Removing a total of 37608 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_dopunrstslt01_250_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38226"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38226
.. number of calphas in PDB: 0
.. number of residues in PDB: 9397
Retaining 938 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Retaining 606 non-hydrogen atoms
Removing a total of 37596 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Retaining 606 non-hydrogen atoms
Removing a total of 37596 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Retaining 606 non-hydrogen atoms
Removing a total of 37596 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Retaining 606 non-hydrogen atoms
Removing a total of 37596 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_noligand_250_restart3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 38202"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 38202
.. number of calphas in PDB: 0
.. number of residues in PDB: 9395
Retaining 914 non-water atoms
Removing a total of 37288 atoms
Renumbering residues ( from 11 ) and atoms ( from 1 )
Warning in convert.pdb(., renumber = TRUE, first.resno = 11, rm.h = FALSE, : Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
The 'cran_repo' argument in shelf() was not set, so it will use
cran_repo = 'https://cran.r-project.org' by default.
To avoid this message, set the 'cran_repo' argument to a CRAN
mirror URL (see https://cran.r-project.org/mirrors.html) or set
'quiet = TRUE'.
── Column specification ────────────────────────────────────────────────────────
cols(
.default = col_character(),
X7 = col_double(),
X16 = col_double(),
X20 = col_double(),
X21 = col_double(),
X22 = col_double(),
X24 = col_double()
)
ℹ Use `spec()` for the full column specifications.
[1] "Reading file D:/Amber/dop/pmemd/out/3_eq_doplong7_5.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 10"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
[1] "Reading file D:/Amber/dop/pmemd/out/3_eq_doplong8_5.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 10"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
[1] "file already exists"
Retaining 1485 non-water atoms
Removing a total of 32166 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
Retaining 1505 non-water atoms
Removing a total of 43839 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Removing a total of 42888 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 5000"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Removing a total of 58452 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_1.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Retaining 975 non-hydrogen atoms
Removing a total of 43398 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Retaining 975 non-hydrogen atoms
Removing a total of 43398 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Retaining 975 non-hydrogen atoms
Removing a total of 43398 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Retaining 975 non-hydrogen atoms
Removing a total of 43398 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_1.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Retaining 995 non-hydrogen atoms
Removing a total of 58962 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Retaining 995 non-hydrogen atoms
Removing a total of 58962 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Retaining 995 non-hydrogen atoms
Removing a total of 58962 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Retaining 995 non-hydrogen atoms
Removing a total of 58962 atoms
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_1.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Removing a total of 42888 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Removing a total of 42888 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Removing a total of 42888 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong7_250_4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 44373"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 44373
.. number of calphas in PDB: 0
.. number of residues in PDB: 10821
Retaining 1485 non-water atoms
Removing a total of 42888 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_1.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Removing a total of 58452 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_2.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Removing a total of 58452 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_3.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Removing a total of 58452 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
[1] "Reading file D:/Amber/dop/pmemd/out/4_prod_doplong8_250_4.nc"
[1] "Produced by program: pmemd"
[1] "File conventions AMBER version 1.0"
[1] "Frames: 6250"
[1] "Atoms: 59957"
Summary of PDB generation:
.. number of atoms in PDB determined by 'xyz'
.. 0 atom(s) from 'string' selection
.. 0 atom(s) in final combined selection
.. number of atoms in PDB: 59957
.. number of calphas in PDB: 0
.. number of residues in PDB: 14732
Retaining 1505 non-water atoms
Removing a total of 58452 atoms
Warning in convert.pdb(., rm.h = FALSE, rm.wat = TRUE): Additional hydrogen elety names may need converting.
N.B. It is often best to remove hydrogen (rm.h=TRUE)
before building systems for simulation
used (Mb) gc trigger (Mb) max used (Mb)
Ncells 2410536 128.8 4015078 214.5 4015078 214.5
Vcells 710089618 5417.6 2942469788 22449.3 3980524038 30369.0
In [16]:
if (!file.exists('images/longrMD.1000frames.pdb')) {write.pdb(pdb = pdb.long,xyz = pdb.long$xyz[seq(1, nrow(pdb.long$xyz), by =nrow(pdb.long$xyz)/1000),],file ='images/longrMD.1000frames.pdb' )} if (!file.exists('images/longuMD.1000frames.pdb')) {write.pdb(pdb = pdb.unrst.long,xyz = pdb.unrst.long$xyz[seq(1, nrow(pdb.unrst.long$xyz), by =nrow(pdb.unrst.long$xyz)/1000),],file ='images/longuMD.1000frames.pdb' )} if (!file.exists('images/longnoLDPMD.1000frames.pdb')) {write.pdb(pdb = pdb.noldp.long,xyz = pdb.noldp.long$xyz[seq(1, nrow(pdb.noldp.long$xyz), by =nrow(pdb.noldp.long$xyz)/1000),],file ='images/longnoLDPMD.1000frames.pdb' )} if (!file.exists('images/longdoplong7.1000frames.pdb')) {write.pdb(pdb = doplong7_longtraj,xyz = doplong7_longtraj$xyz[seq(1, nrow(doplong7_longtraj$xyz), by =nrow(doplong7_longtraj$xyz)/1000),],file ='images/longdoplong7.1000frames.pdb' )} if (!file.exists('images/longdoplong8.1000frames.pdb')) {write.pdb(pdb = doplong8_longtraj,xyz = doplong8_longtraj$xyz[seq(1, nrow(doplong8_longtraj$xyz), by =nrow(doplong8_longtraj$xyz)/1000),],file ='images/longdoplong8.1000frames.pdb' )}
In [17]:
.justify {text-align:justify!important}
1 Methods
1.1 System preparation
The system was prepared using the leap program from the Amber24 suite (Case et al. 2023). The OL21 and AMBER General Force Field for organic molecules (Version 1.81) were used to describe the complex (J. Wang et al. 2004; Zgarbová, Šponer, and Jurečka 2021; Love et al. 2023). The structure was explicitly solvated in a truncated octahedral box of water molecules, using the OPC model with the ad hoc Li/Merz ion parameters of atomic ions (12-6 set)(Izadi, Anandakrishnan, and Onufriev 2014; Li et al. 2020), with a minimum of 14 Å between the solute and the box edge. For simulations in absence of salts (case of unrestrained simulations), the system was neutralized by adding 25 Na+ counter-cations. In cases where 140 mM NaCl were added to better reflect the SELEX conditions (restrained simulations), the number of required ions (\(N_{\pm}\); here around 35 Na+ and 10 Cl-) was determined following the SLTCAP method (Schmit et al. 2018), using Equation 1 simplified by Machado and Pantano into Equation 2, where \(\nu_w\) is the water volume of the simulation box (around \(3\times 10^5\) Å3 here) in reduced units, \(c_0\) the salt concentration, \(Q\) the total charge of the complex (here -25: 26 phosphates on the aptamer and 1 ammonium group on the dopamine), and \(N_0 = \frac{N_w \times c_0}{55.5}\), with \(N_w\). the number of water molecules in the simulation box (here, around \(9\times10^4\)) (Machado and Pantano 2020).
Note that the SPLIT method describe by Machado and Pantano cannot be applied as our system does not satisfy the \(N_0 \gg Q\) condition; however it yields identical values in most cases or deviate by a single ion.
1.2 Restrained minimization, heating and molecular dynamics
Using an in-house R script (R Core Team 2023; Wickham et al. 2019), the distance restraints obtained in ARIA were converted to an 8-column format suitable for its processing by the makeDIST_RST function from the sander module of Amber. A custom map file was prepared to define common names for groups of protons sharing a given restraints (e.g. 3 H from a same methyl group). The resulting DISANG restraints file was then applied to all steps below.
All simulation steps were performed with pmemd.cuda (v. 18.0.0) from the CUDA version of AMBER (Götz et al. 2012; Salomon-Ferrer et al. 2013; Le Grand, Götz, and Walker 2013), on an NVIDIA H100 PCIe Tensor core GPU (CUDA version: 12.4) from the DOREMI CALI v3 cluster of the Mésocentre de Calcul Intensif Aquitain (Université de Bordeaux). The system was minimized for 20000 cycles using the steepest descent algorithm for the first 4000 steps and the conjugate gradient for the next 16000 steps. The weights of the restraints were kept constant at 100 kcal.mol-1.Å-2. The system was then heated at constant volume from 0 to 298 K over 18 ps then kept for 2 ps at the final temperature, using a time step of 2 fs, the Langevin thermostat with a 2.0 ps-1 collision frequency and a different seed for the pseudo-random number generation for every run to avoid synchronization artifacts (Sindhikara et al. 2009), an 8 Å non-bonded cutoff, and the bonds involving hydrogen were constrained with the SHAKE algorithm. The system was further equilibrated five times at 298 K with the parameters above and the restraint weights ramping down from 100 to 5 kcal.mol-1.Å-2. The pressure was kept at 1 bar with the Berendsen barostat (Berendsen et al. 1984).
Restrained molecular dynamics for subsequent minimization were ran for 10 ns with the parameters above, and restraints weights set at 20 kcal.mol-1.Å-2. The final coordinates were further minimized, as described above except for the restraint weights set at 20 kcal.mol-1.Å-2. Remaining NMR violations were summarized with the sviol function from the Amber package.
Production MD were run over a microsecond with and without restraints, as well as in absence of dopamine, with the parameters above.
1.3 Data analysis
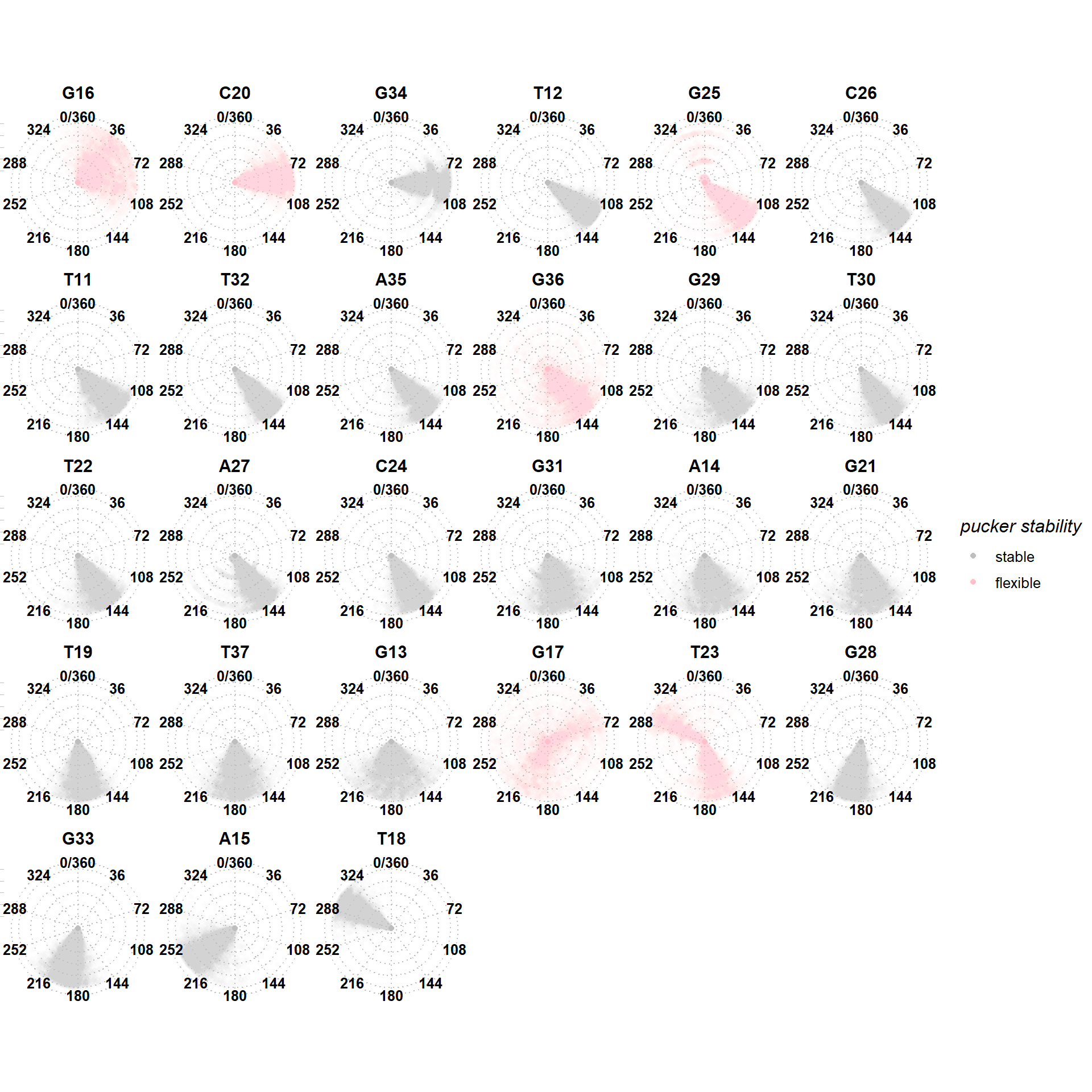
The bio3d package was used for minimized structure and trajectory files cleanup, alignment, filtering, averaging and analysis (B. J. et al. 2006). The determination of RMSD and RMSF was performed with the rmsd and rmsf functions. Dihedral angles, sugar pucker angles and amplitudes \(\theta_M\) were obtained with in-house R functions leveraging the torsion.xyz function, following Equation 3, where the pucker P is determined by Equation 4 and the sugar torsion angles \(\nu_i\) are defined by four atoms as shown below.
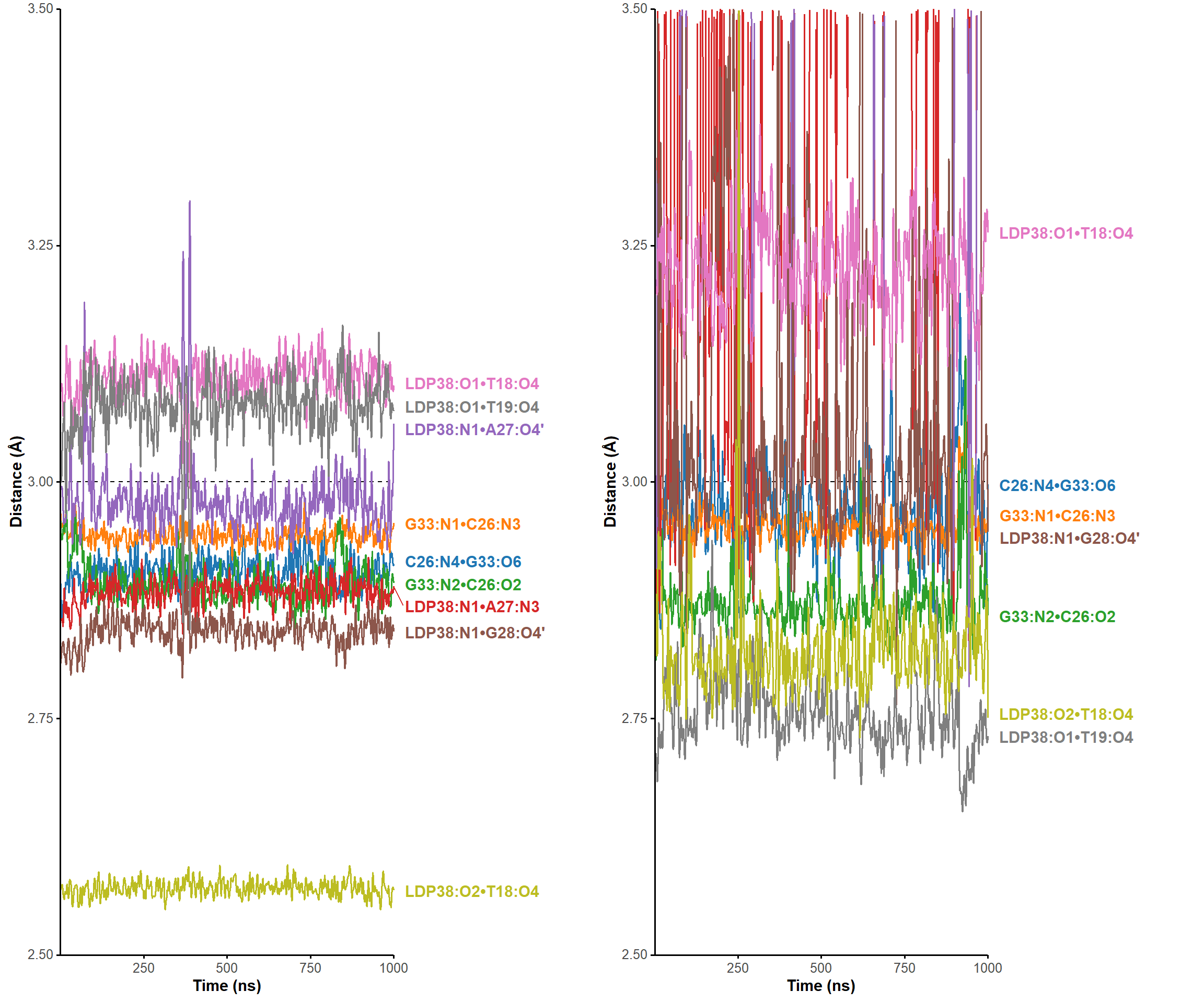
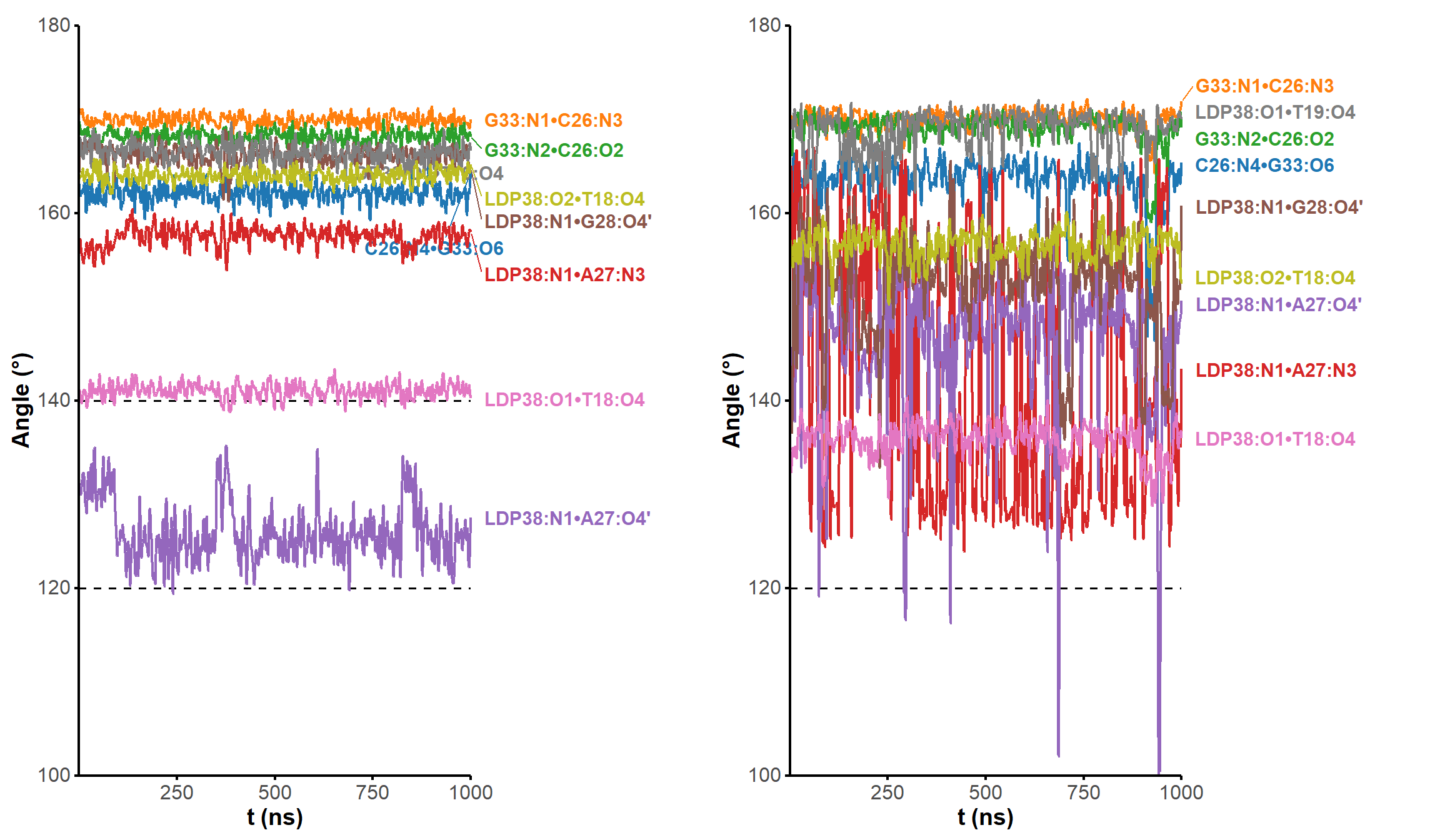
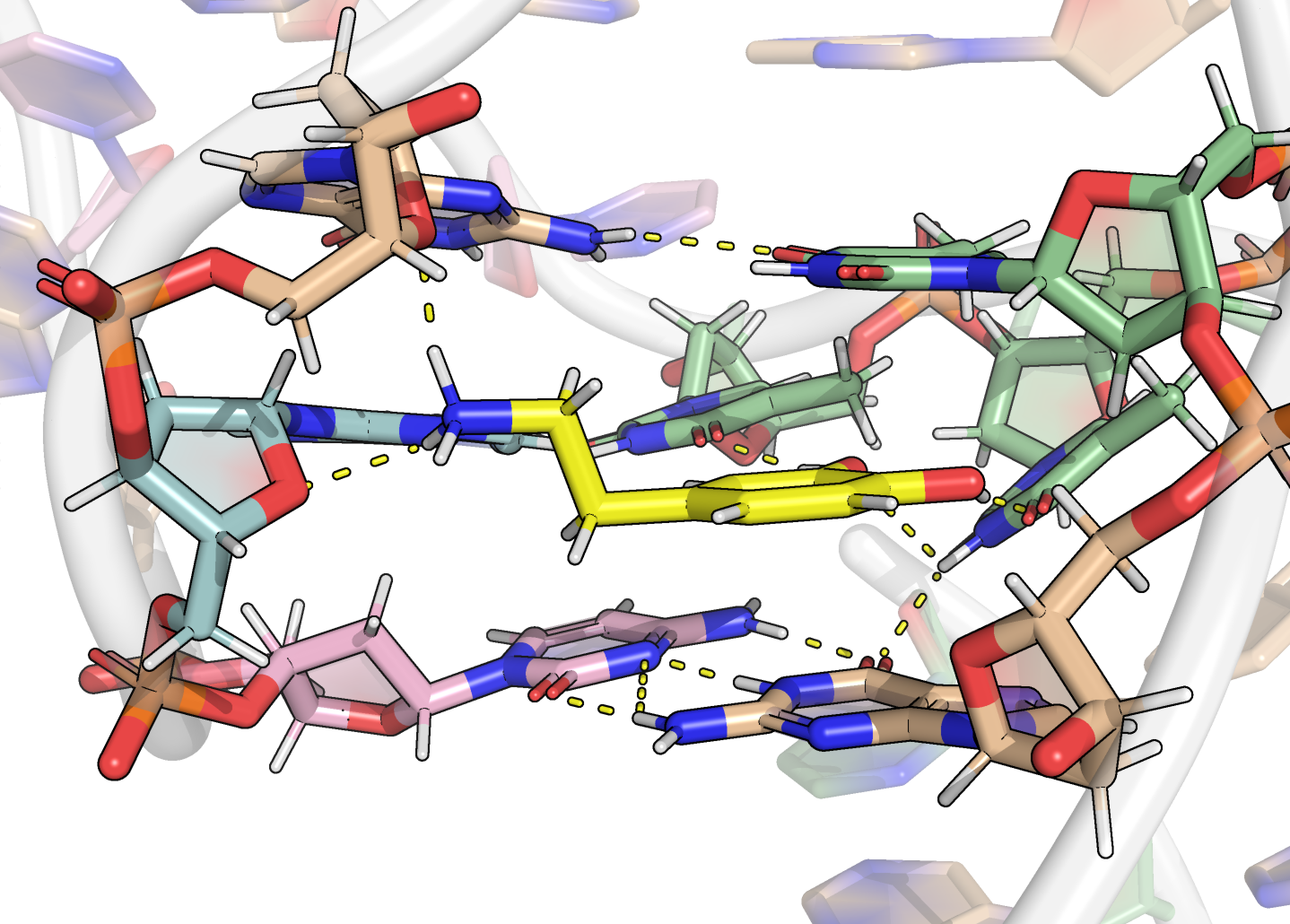
All atom-atom and ring-ring distances and angles were measured with the dist.xyz and angle.xyz functions. In-house R scripts were used to infer the formation of H-bonds from these values on full trajectories, by first selecting realistic donor/acceptor atoms for each residue/ligand, then verifying that the donor/acceptor distances and angles were compatible with H-bond formation.
Principal component analysis on the final structures and trajectories (all atoms except hydrogen) was performed with the pca function from bio3d. The results were clustered with the k-means method using Euclidean distance and the best number of clusters determined by Nbclust(Charrad et al. 2014). Cross-correlation analysis was carried out with the dccm function of bio3d.
Molecular structures images were created in PyMOL 2.5 (Schrödinger, LLC 2021), interactive molecular structures with mol*, and ligand binding site diagram with LigPlot+(Laskowski and Swindells 2011). All further data processing and all plotting was performed in R 4.3. Apache Arrow was used to write/read processed data files in feather format (Richardson et al. 2024).
2 Results
Hereafter, are described the results for the restrained simulations over 10 ns for the fifteen starting structures, the first of which was also simulated for a microsecond. As a control, the latter was also performed in absernce of restraints.
2.1 Trajectory
2.1.1 Visualization
2.1.1.1 Microsecond simulation
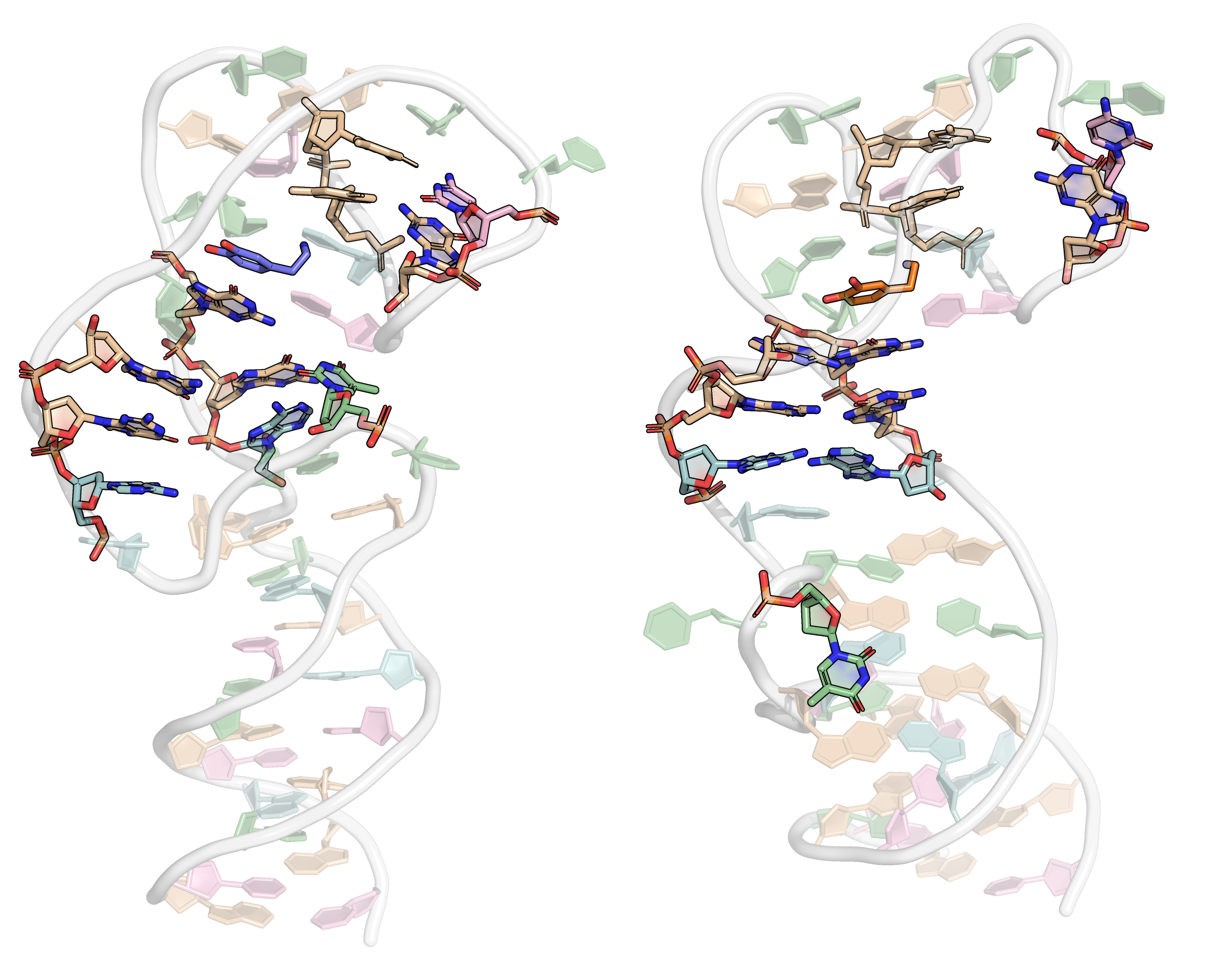
Below can be seen a comparison between the restrained and unrestrained microsecond simulations. One frame every 10 ns was extracted.
2.1.1.2 Production simulation for minimization
All twenty restrained molecular dynamic trajectories (10 ns) can be visualized below. Only one every 5 frames were extracted. Click on the top-left play button to animate the trajectory.
2.1.2 Average structures
In [18]:
# split pdb.long$xyz into 20 matrices of n.frames/20 rowsmean.xyz.list <-split(as.data.table(pdb.long$xyz), rep(1:20, each =nrow(pdb.long$xyz) %/%20))# average each coordinate (per atom/dimension) for each matrixmean.xyz <-lapply(mean.xyz.list, function(x) as.numeric(colMeans(x)))# Collapse all numerics into a single matrix of 20 sets of coordinatesmean.xyz <-matrix(unlist(mean.xyz), nrow =20, byrow =TRUE)if (!file.exists('images/mean_long_rMD.pdb')) {write.pdb(pdb = pdb.long, xyz = mean.xyz, file ='images/mean_long_rMD.pdb')}###
Twenty structures of the microsecond restrained simulation were averaged over 1250 frames of the simulation, without further minimization. It is not incredibly useful as it was not further minimized, but it can be done. The minimized structures can be found in Section 2.2.
2.1.3 RMSD
2.1.3.1 Restrained simulations
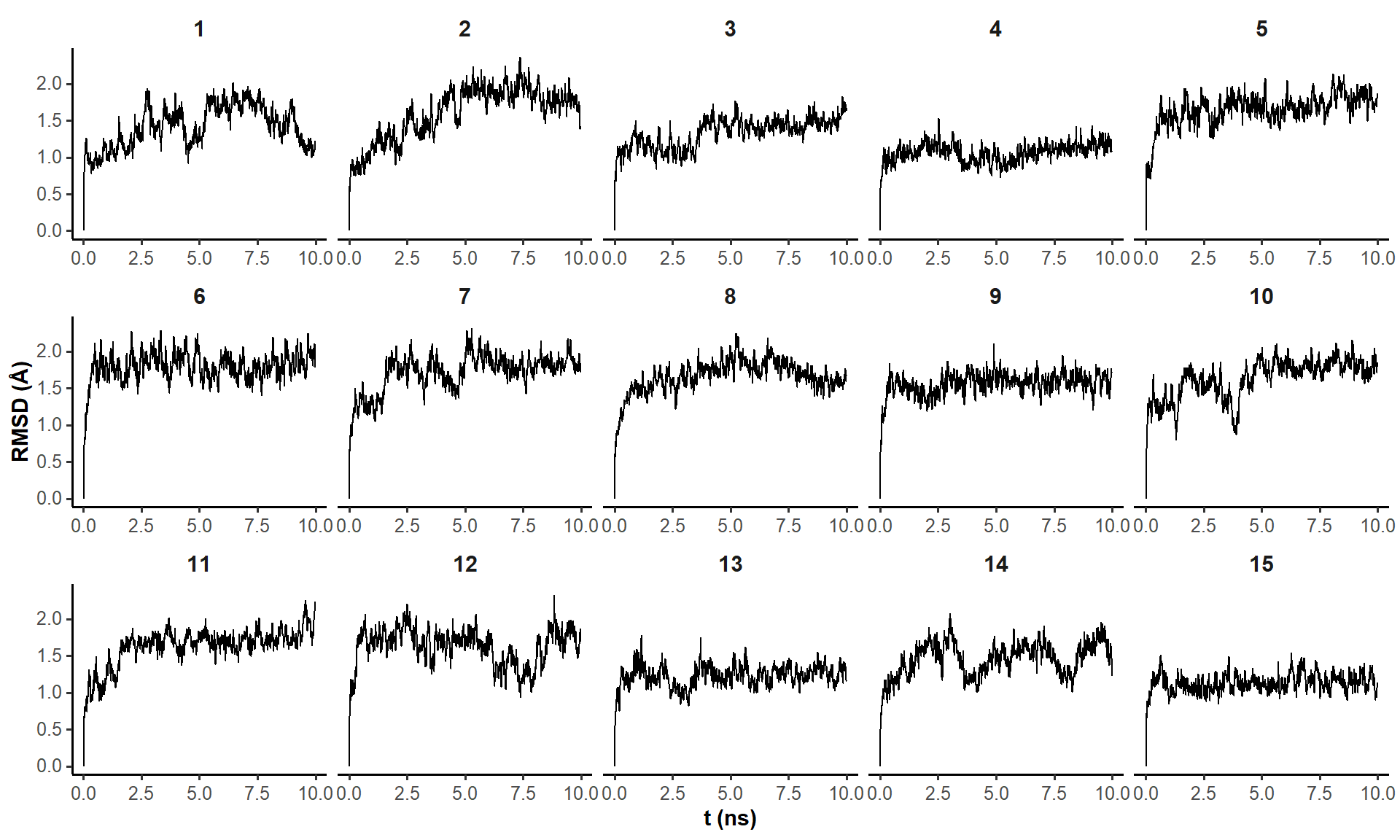
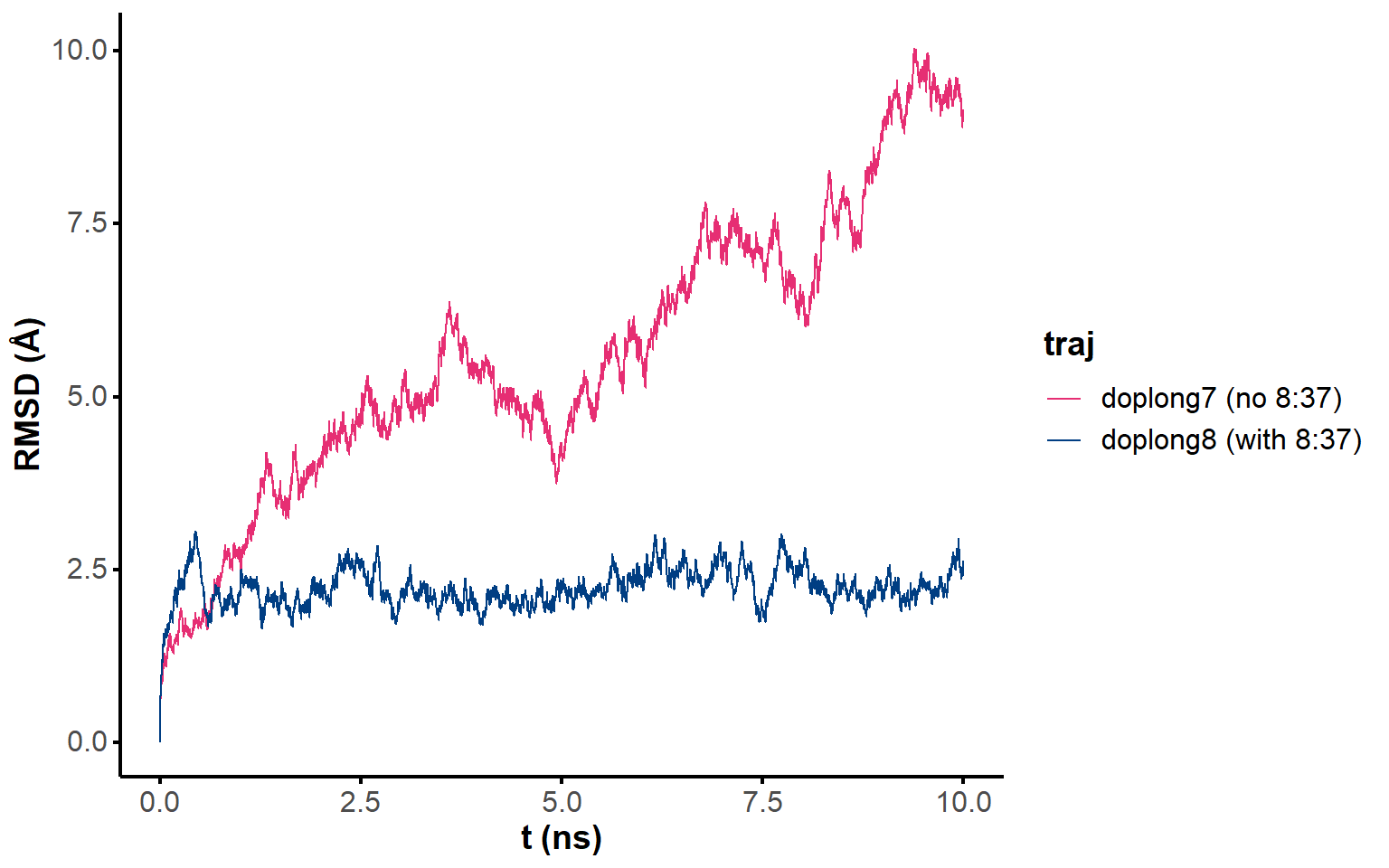
The trajectory of almost all fifteen simulations seem to converge after the first hundreds of ps (Figure 1). Only structure 2 requires around 5 ns to converge. Minimization of the coordinates after ten nanoseconds of simulation is appropriate.
In [19]:
#apply rmsd on all objects of xyz.listrmsd.list <-lapply(1:15, function(i) {rmsd(a = xyz.list[[i]][1,],b = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by =5),],fit =FALSE#already aligned at the import stage )})# bind rows of rmsd.list to make a data.frame, and add a column for the frame number, and a column for the list numberrmsd.df <-do.call(rbind, lapply(1:15, function(i) {data.frame(frame =seq(1, nrow(xyz.list[[i]]), by =5),rmsd = rmsd.list[[i]],structure = i ) })) %>%mutate(t = frame * info.short$time.per.frame)#plot rmsdsrmsd.df %>%ggplot(aes(x = t, y = rmsd)) +geom_line() +custom.theme(0.8) +facet_wrap(~structure,ncol =5,scales ='free_x') +# scale_x_continuous(n.breaks = 3) +labs(x ='t (ns)',y ='RMSD (Å)', )
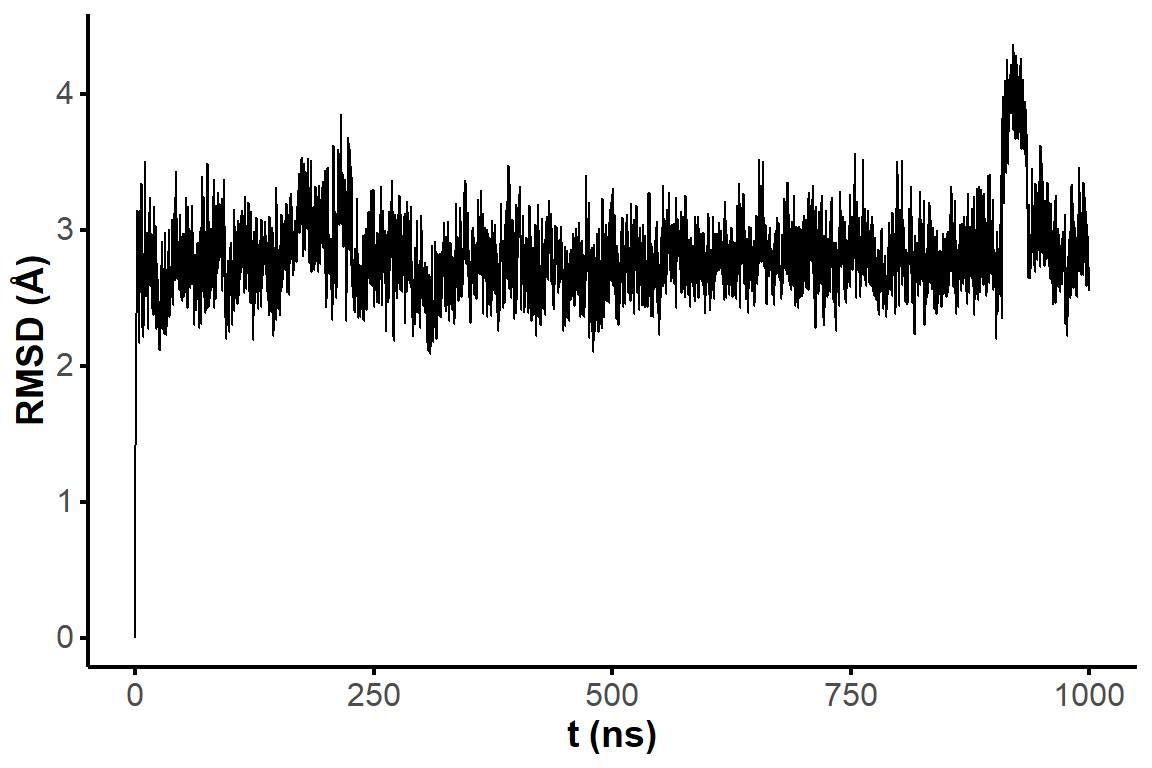
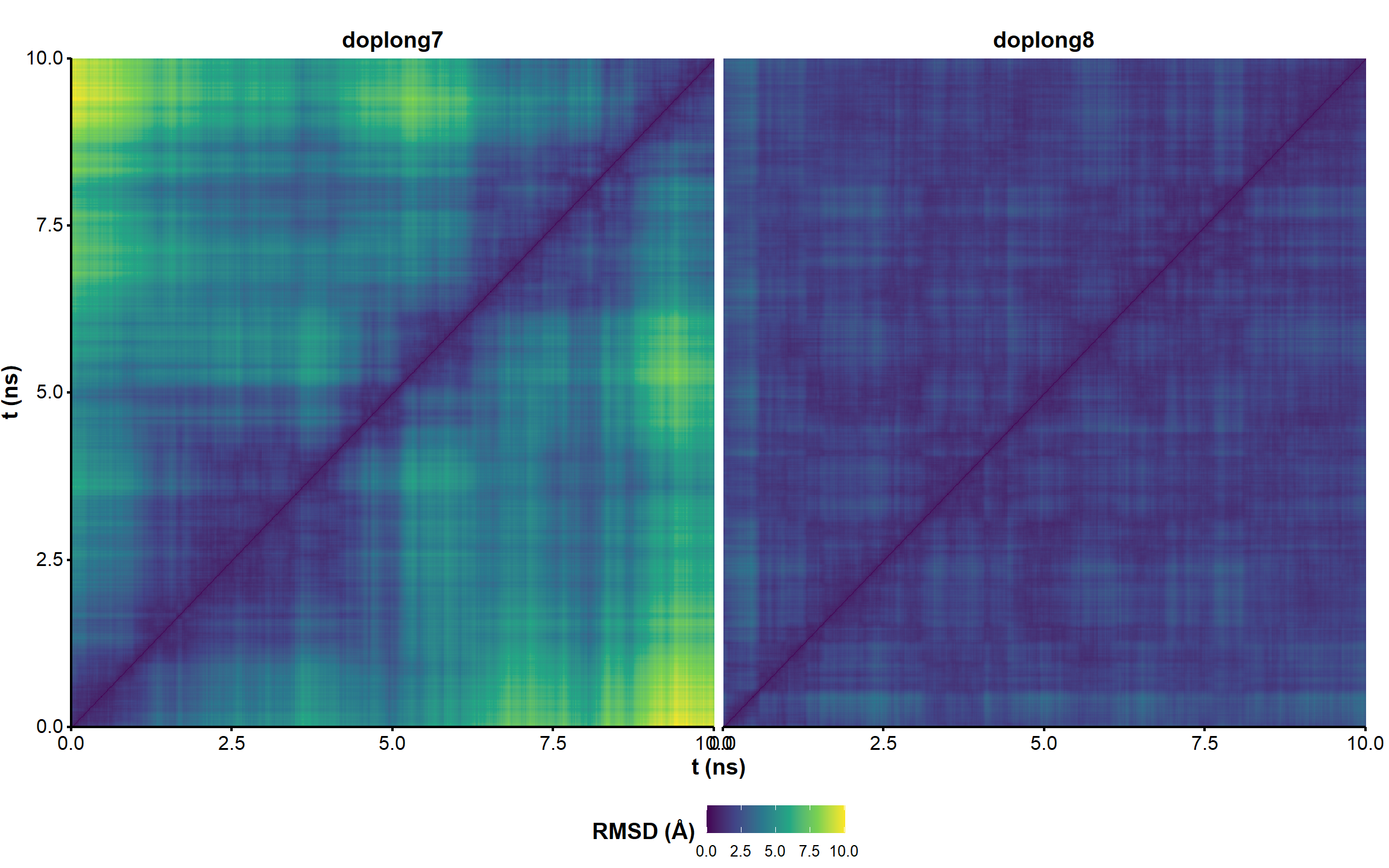
Figure 1: RMSD on all residues (including 5’ and 3’-ends) without hydrogens across the restrained simulations (10 ns), where the first frame is used as reference.
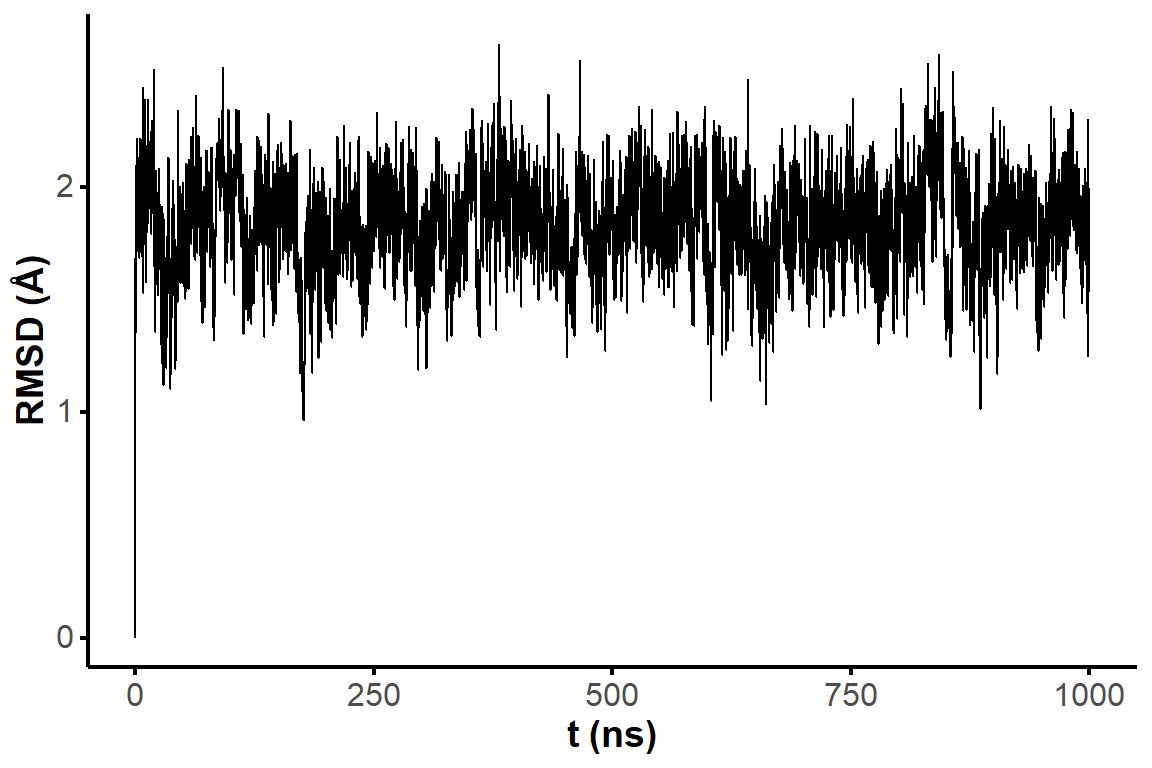
The microsecond simulation has a very stable RMSD vs. the first frame.
Figure 2: RMSD on all residues (including 5’ and 3’-ends) without hydrogens across the restrained simulation (1 microsecond), where the first frame is used as reference.
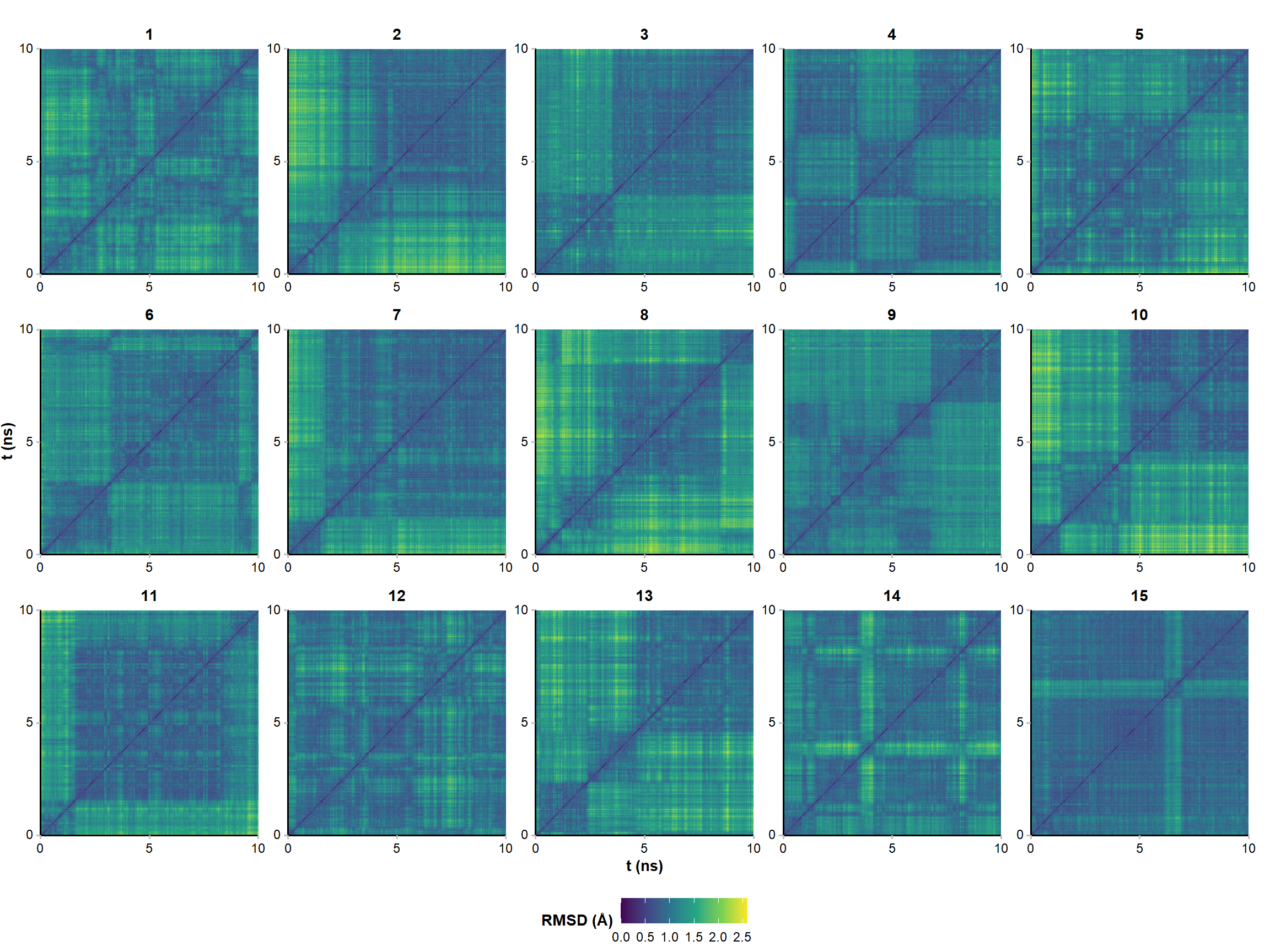
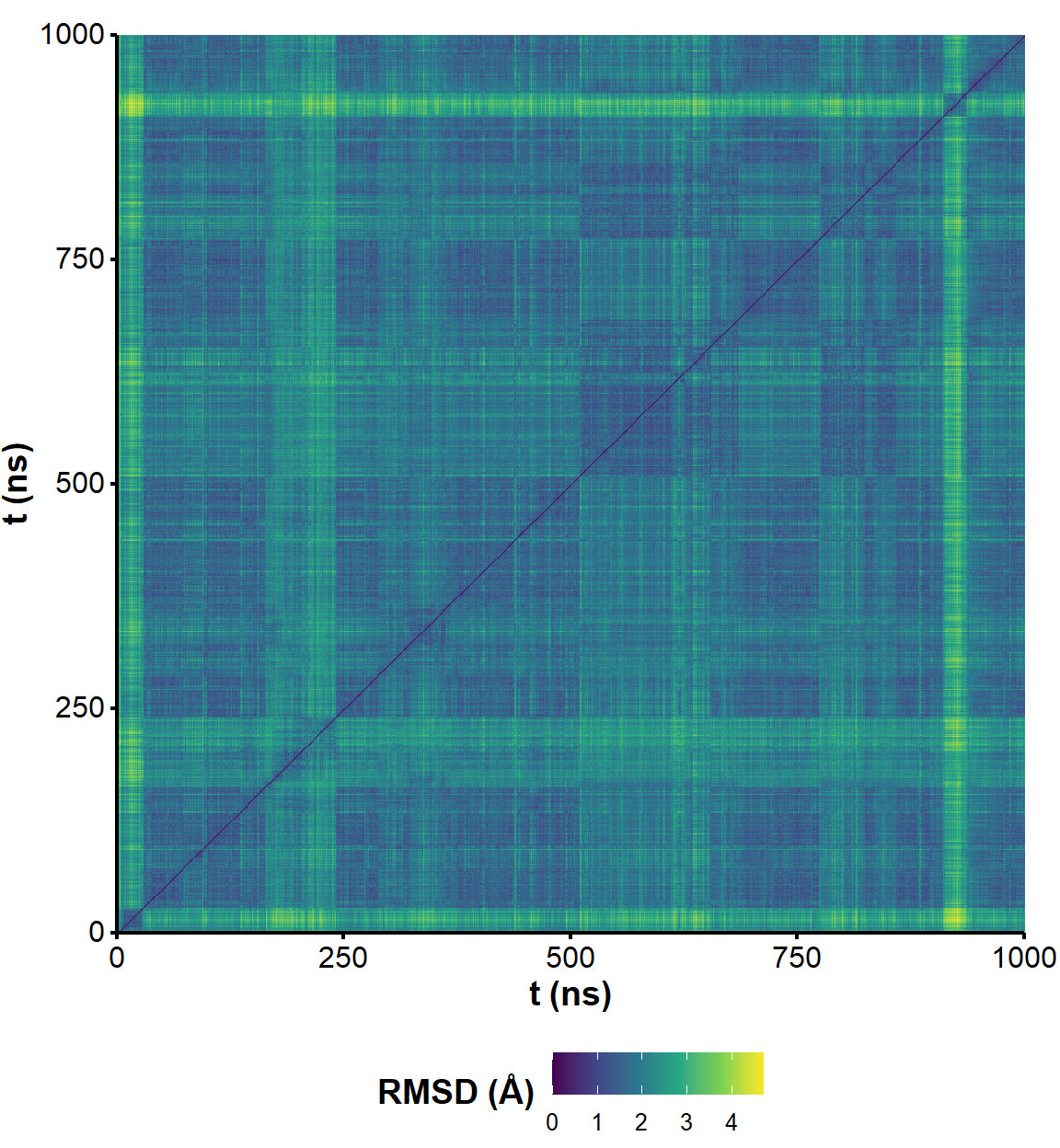
Two frames with similar one-dimensional RMSD are not necessarily similar. Pairwise RMSD better captures the extent of structural diversity across the simulation and avoids the bias from comparing all frames to a single (first) conformer. A few clusters of structures are visible in the RMSD matrices, suggesting that the simulations have converged to a few distinct states (Figure 3). However, there are no very significant variations along the trajectories (RMSD never exceeds ~ 2 Å). The nature of the visited states is investigated for the structure 1 in the sections below.
In [21]:
#calculate only for 1000 framesrmsd.pairwise <-lapply(1:15, function(i) {rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by =5),], #keep one in every 5 framesfit =FALSE ) %>%lazy_dt() %>%mutate(var1 =1:nrow(.), .before =1) %>%pivot_longer(cols =-var1, names_to ="var2", values_to ="value") %>%mutate(var2 =as.numeric(gsub("V", "", var2)),structure = i) %>%as.data.table()}) %>%rbindlist()
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
Warning in rmsd(a = xyz.list[[i]][seq(1, nrow(xyz.list[[i]]), by = 5), ], : No indices provided, using the 571 non NA positions
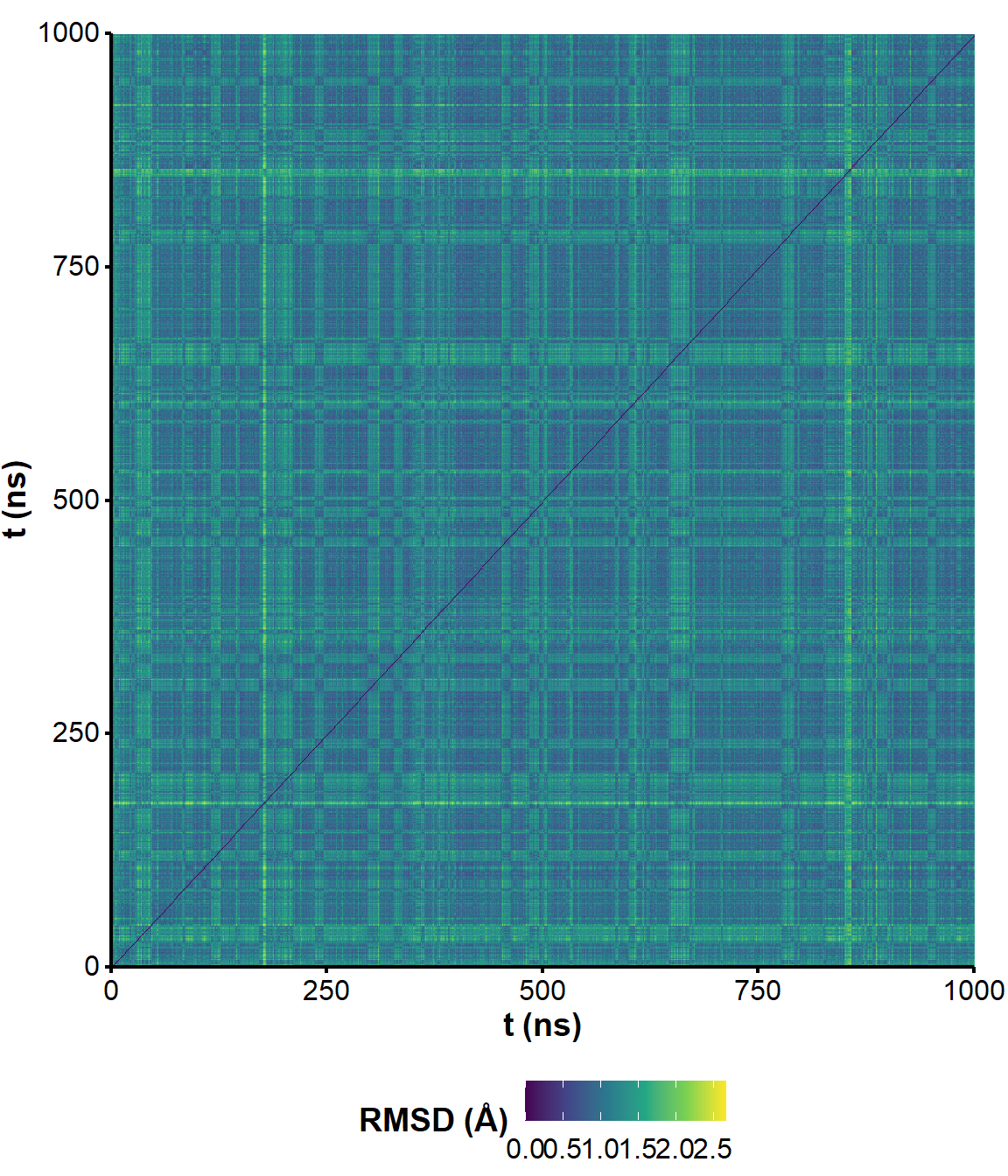
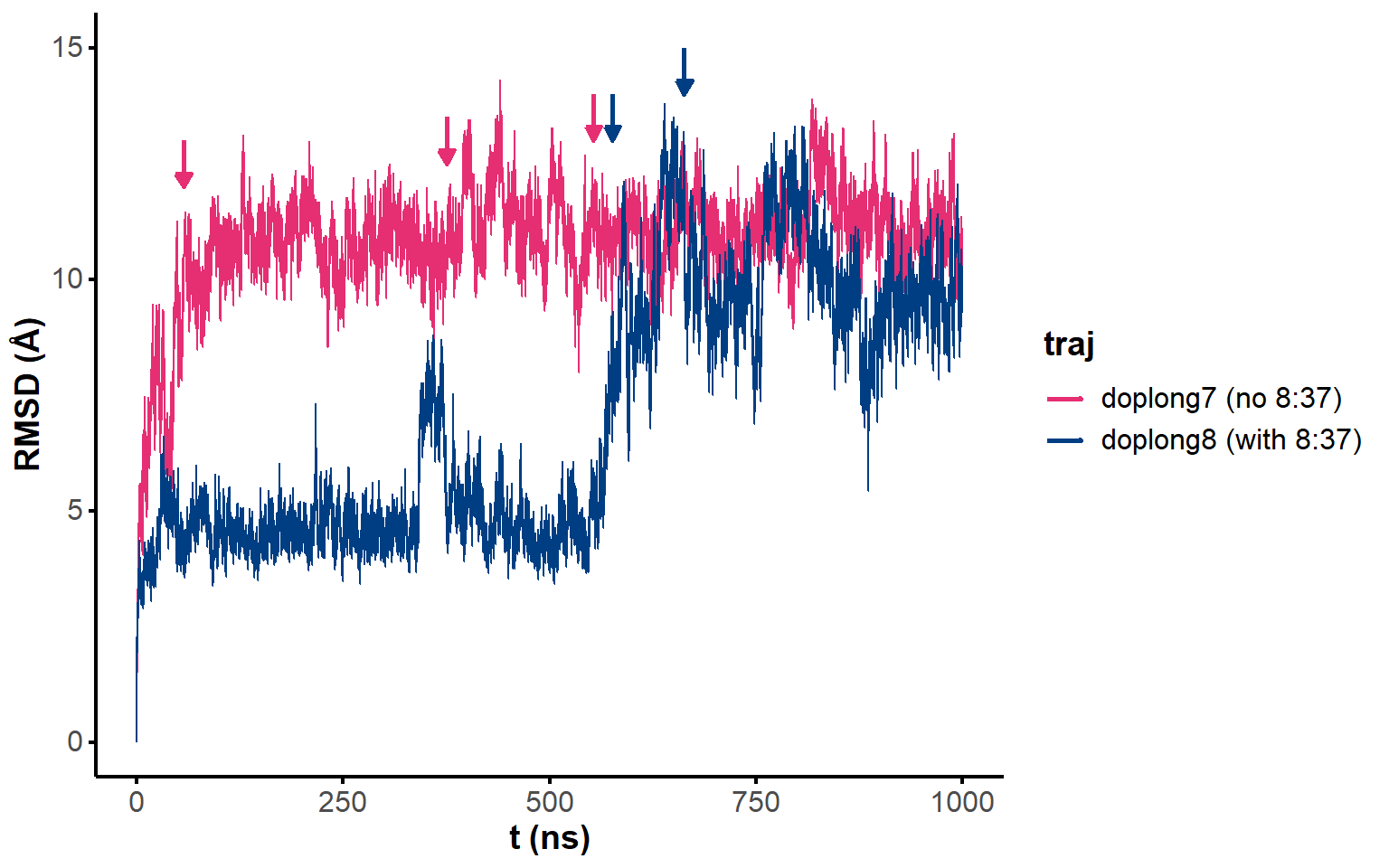
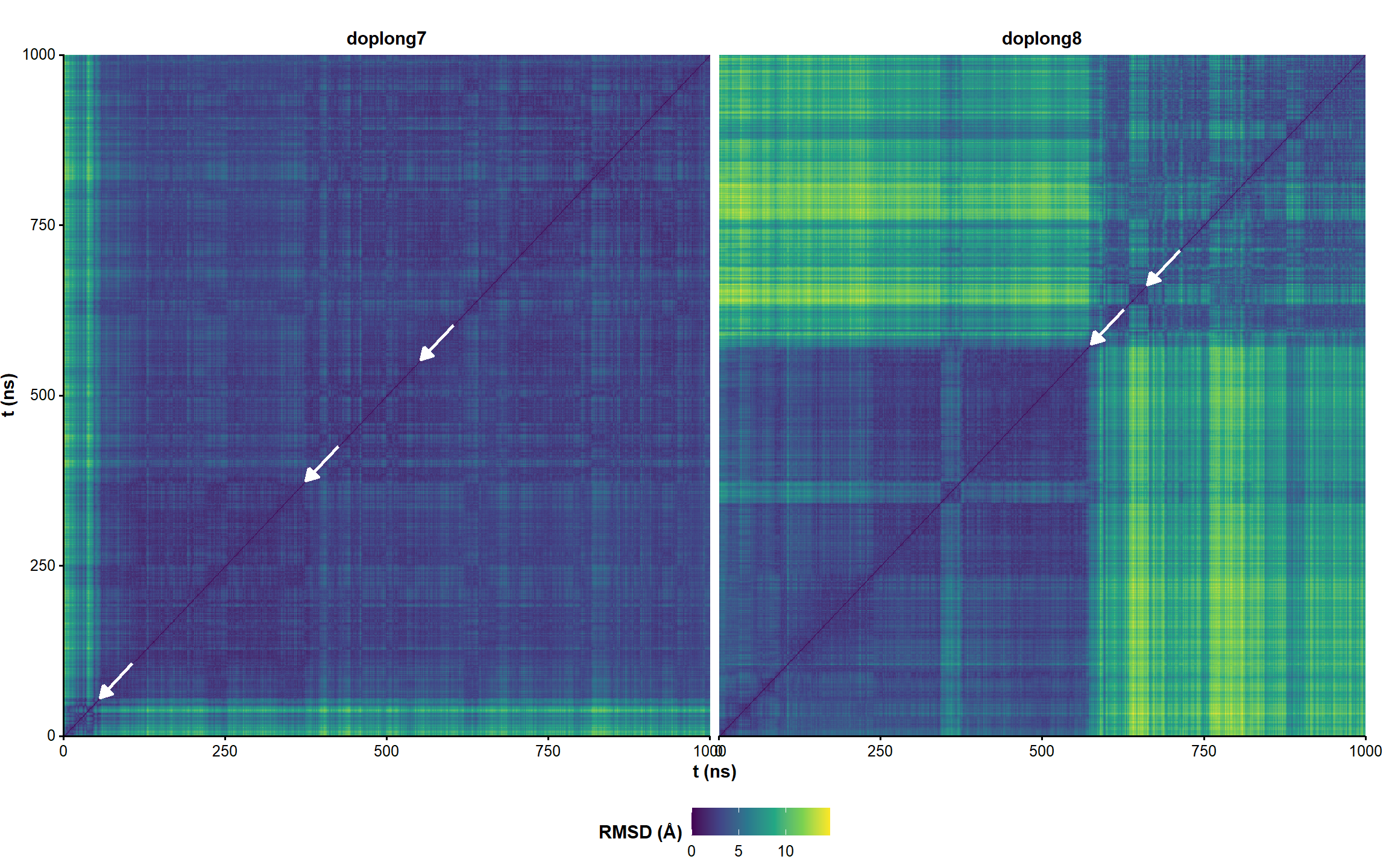
Figure 5: RMSD on all residues (including 5’ and 3’-ends) without hydrogens across the unrestrained simulation, where the first frame is used as reference.
Figure 6: All-to-all RMSD (all residues and ligand atoms except H) of the unrestrained simulation
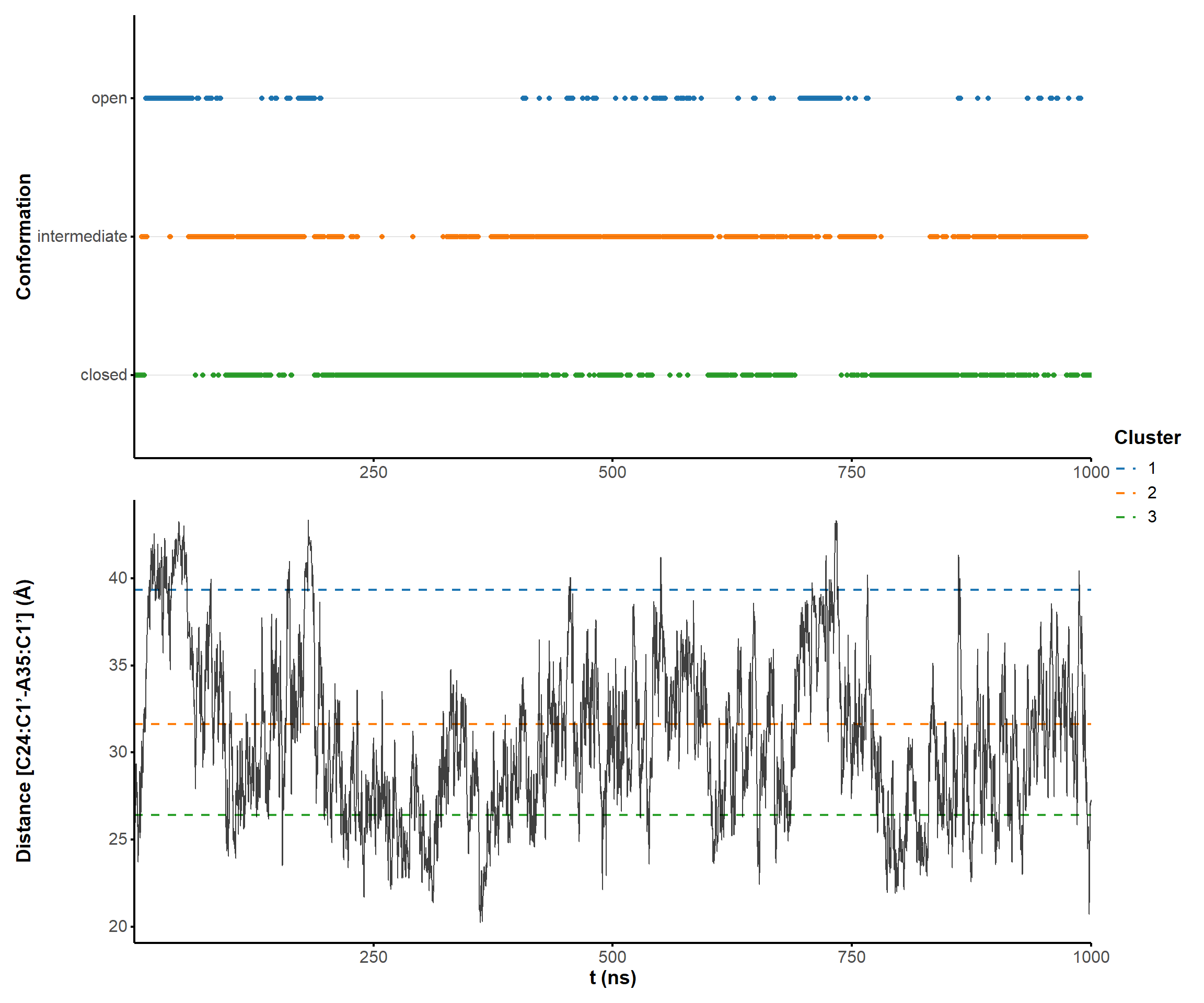
2.1.4 PCA
Given the RMSD heatmaps shown above, it is likely that the aptamer visits different conformations. To isolate discrete states from the microsecond simulation, principal component analysis was performed.
In [28]:
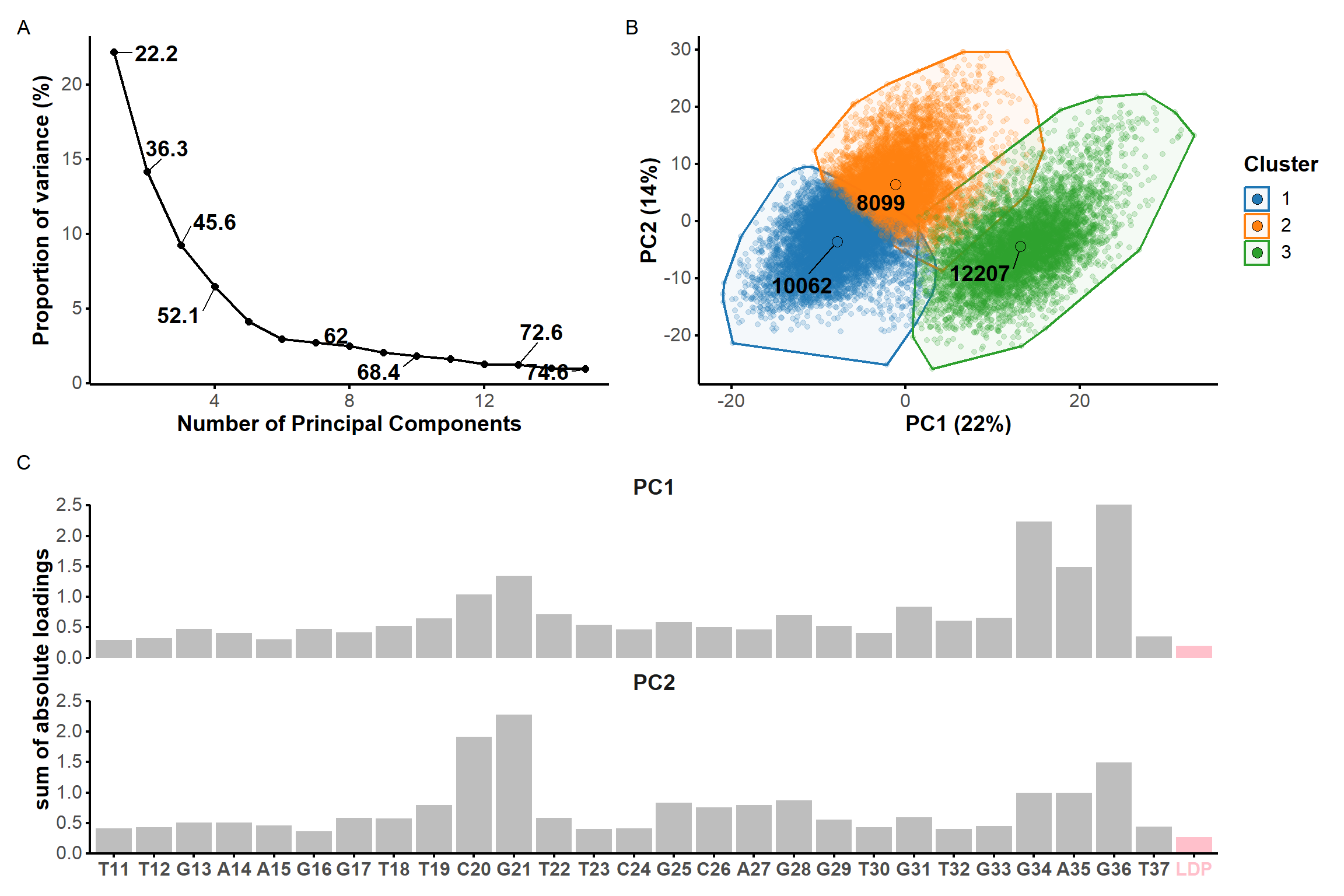
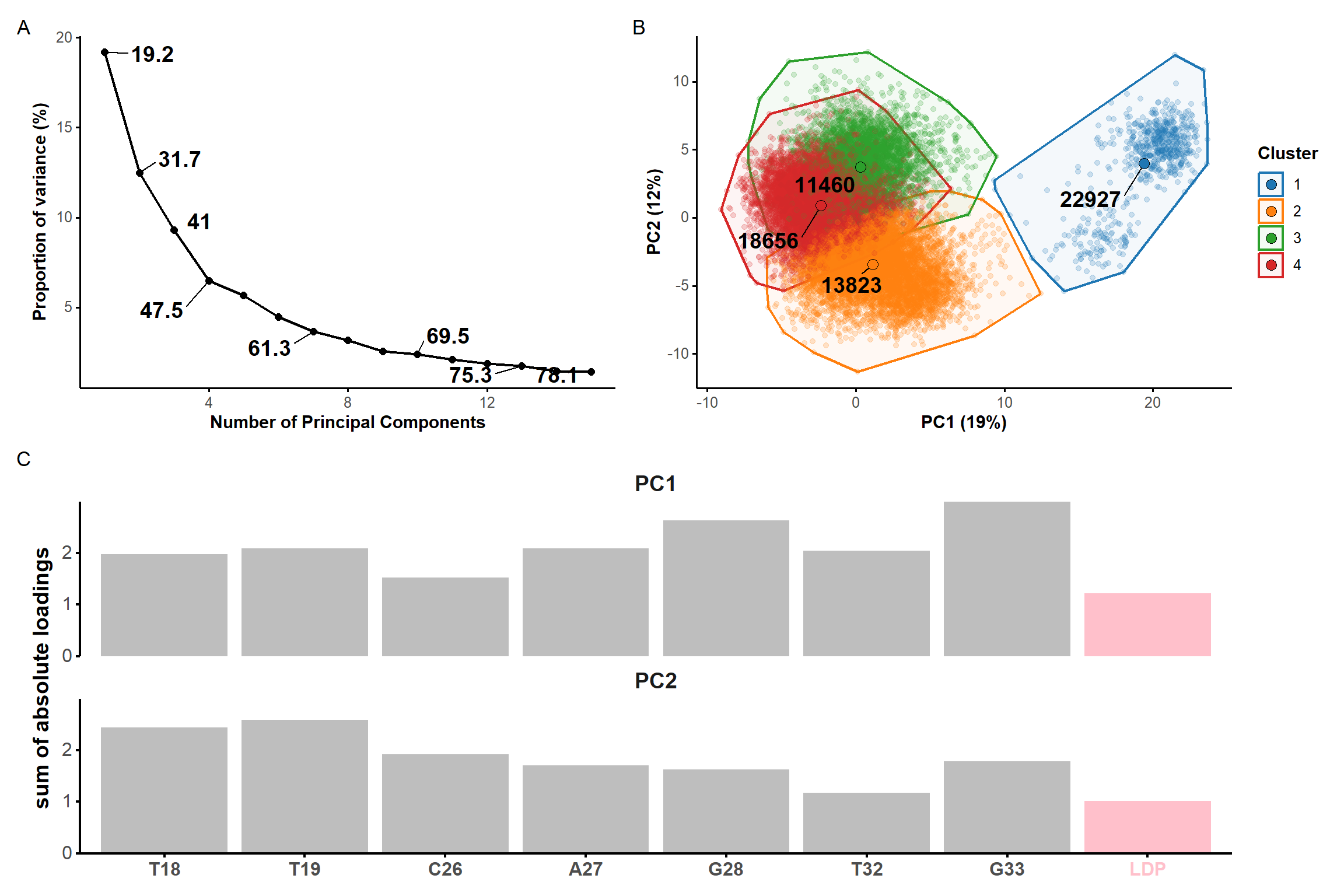
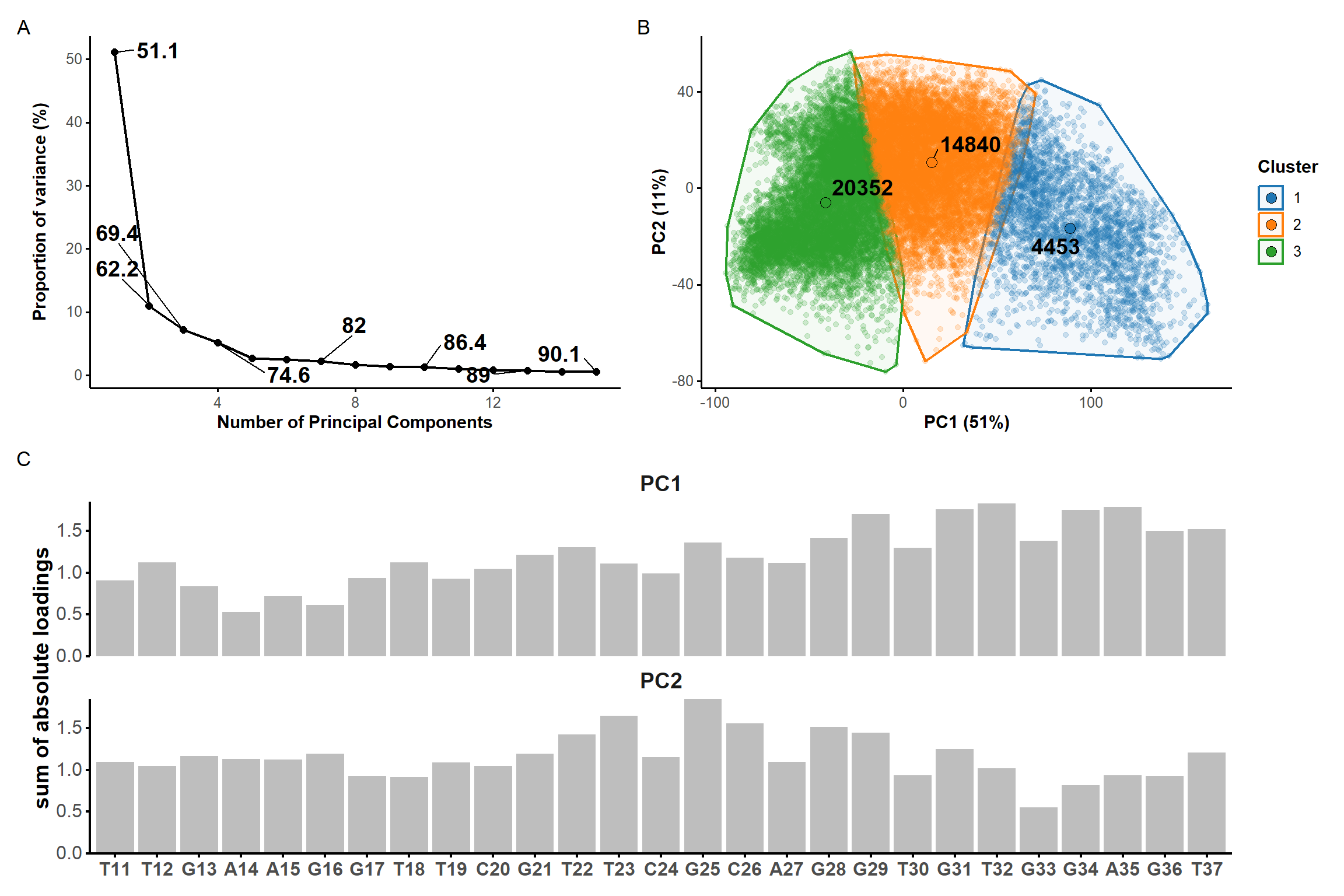
#PCApca_pdb_traj <-pca.pdbs( pdb.long, use.svd =FALSE, rm.gaps =TRUE, fit =FALSE#pdb models are already aligned at the import stage)scree <-data.frame(pc =1:length(pca_pdb_traj$L),L = pca_pdb_traj$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.), signif(cum.var, 3), NA) ) %>%ggplot(., aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' ) ###
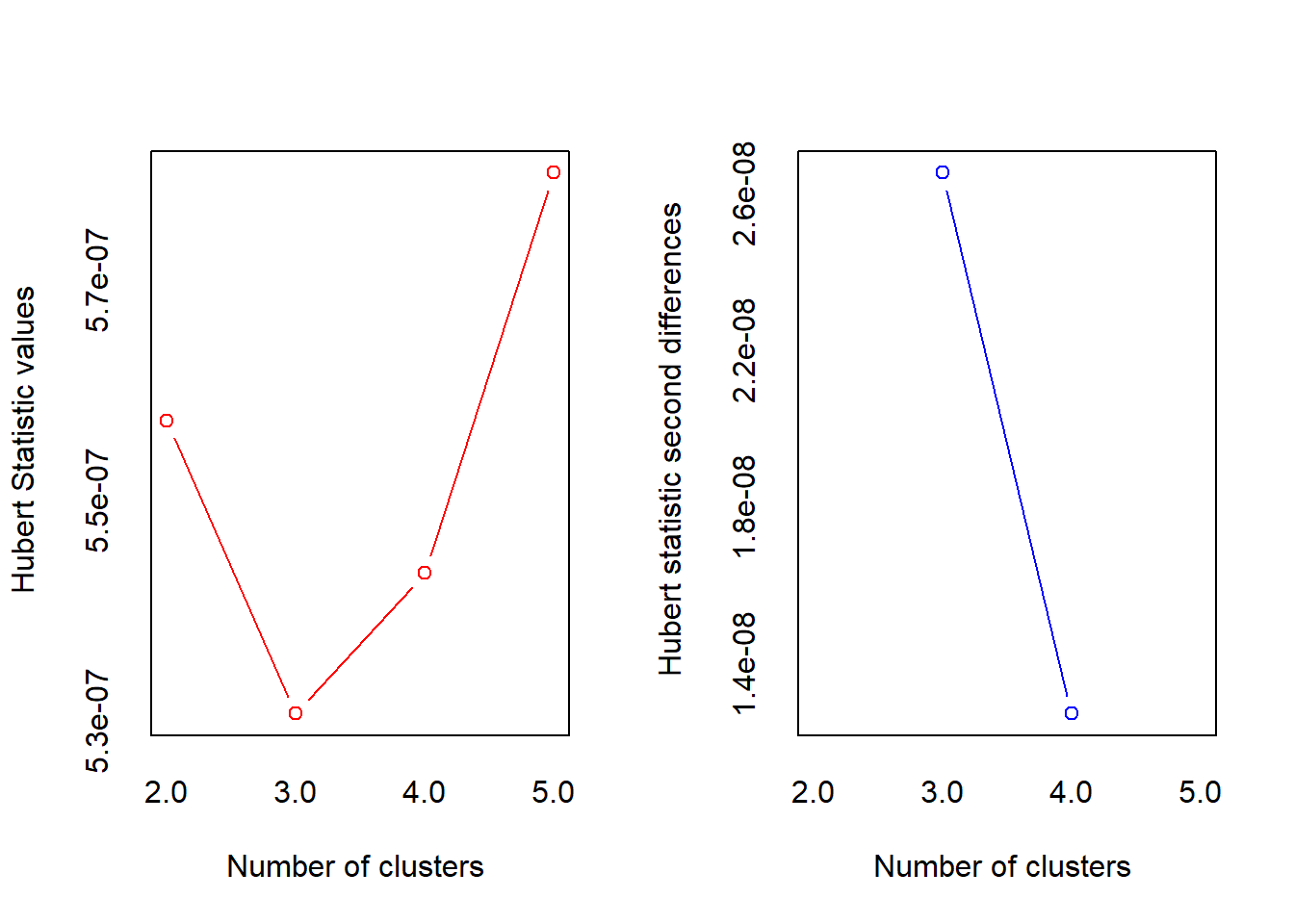
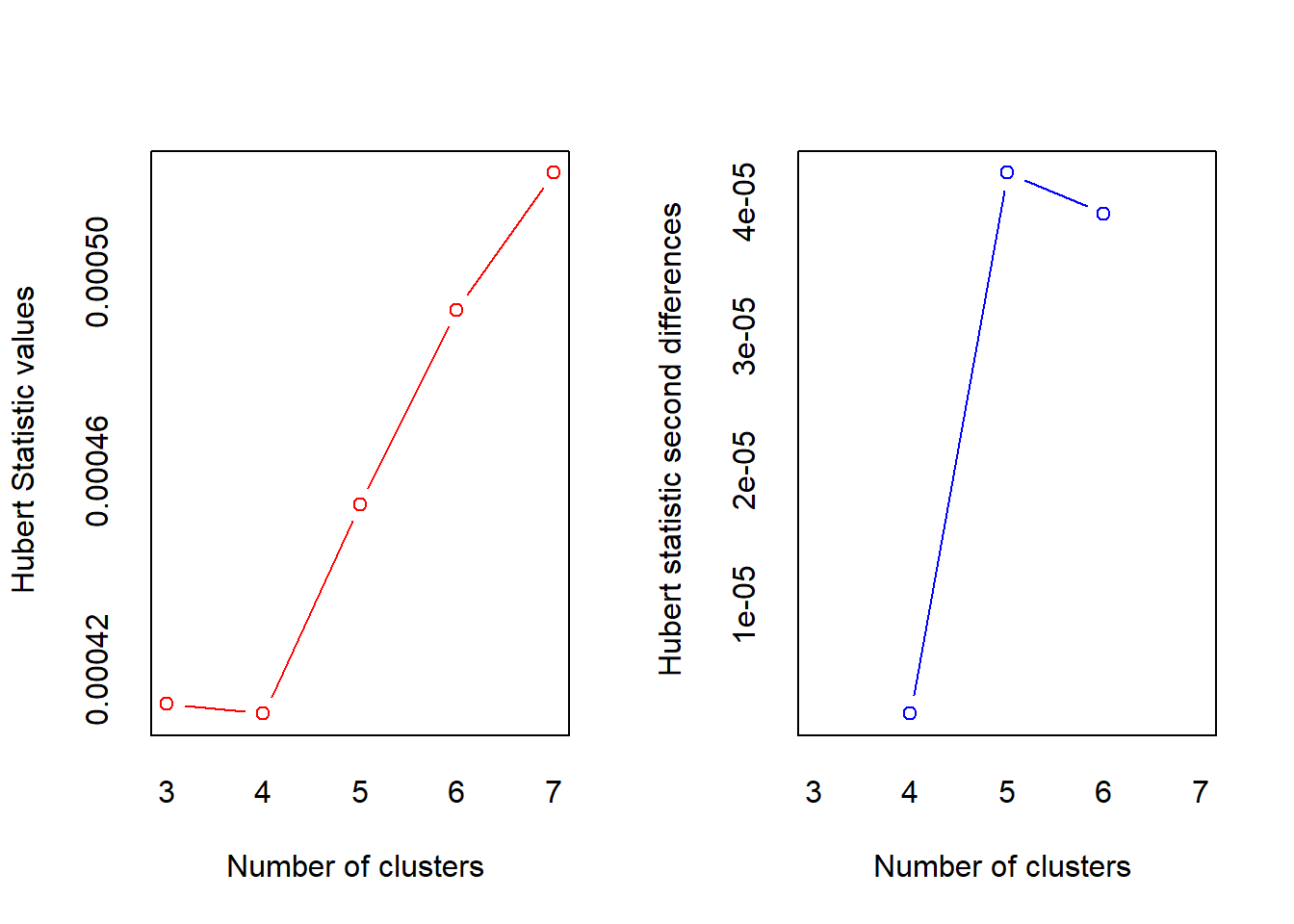
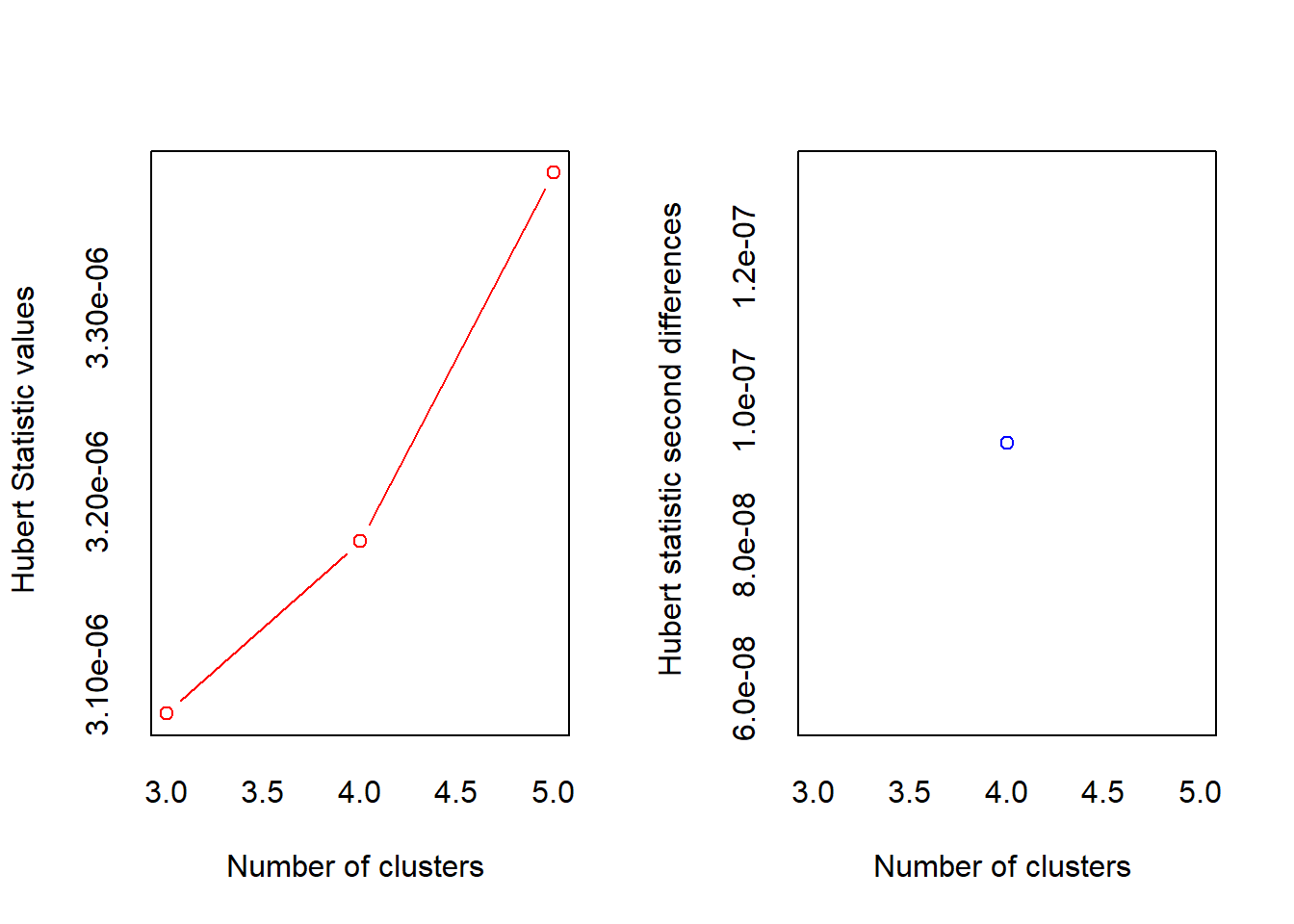
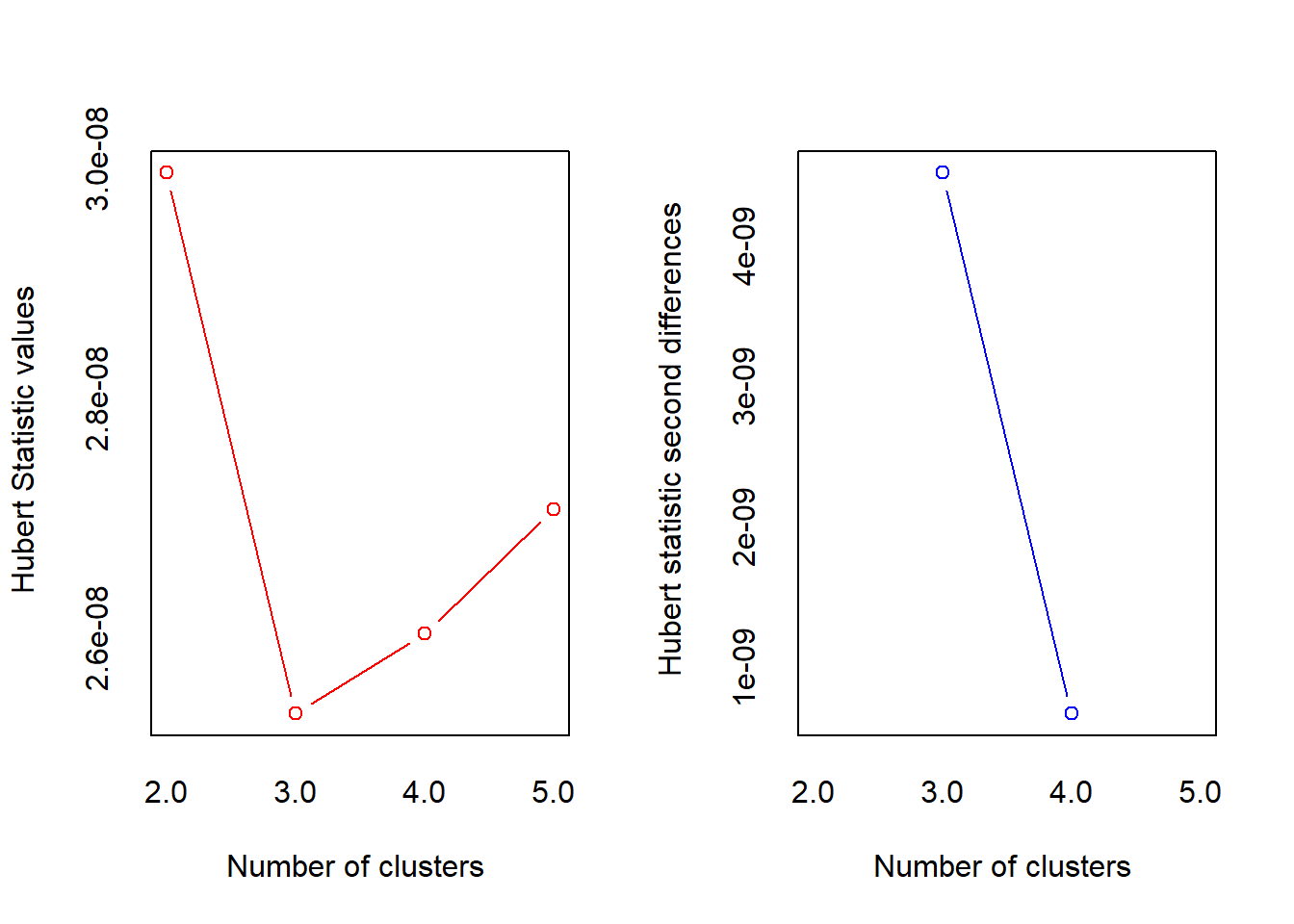
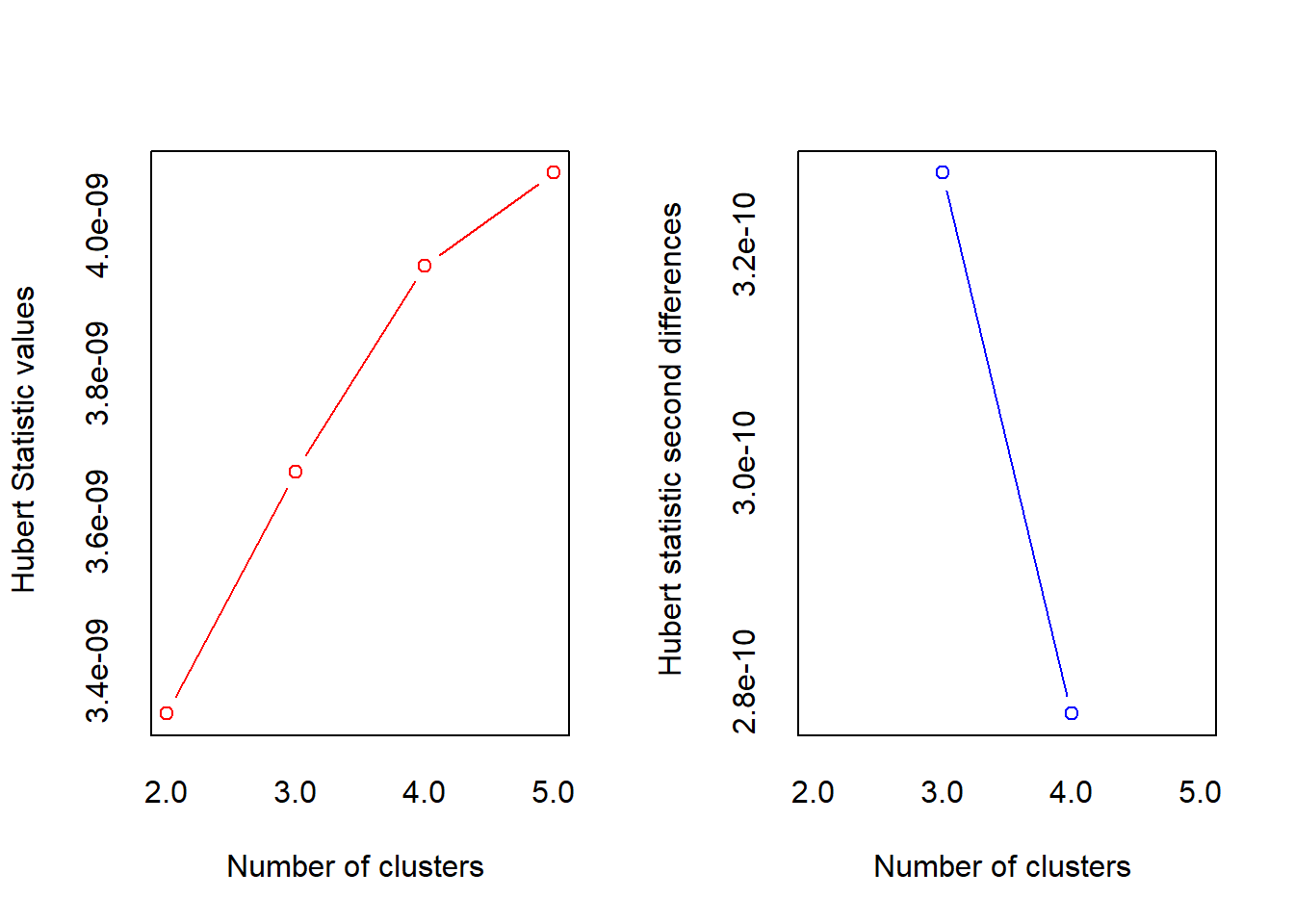
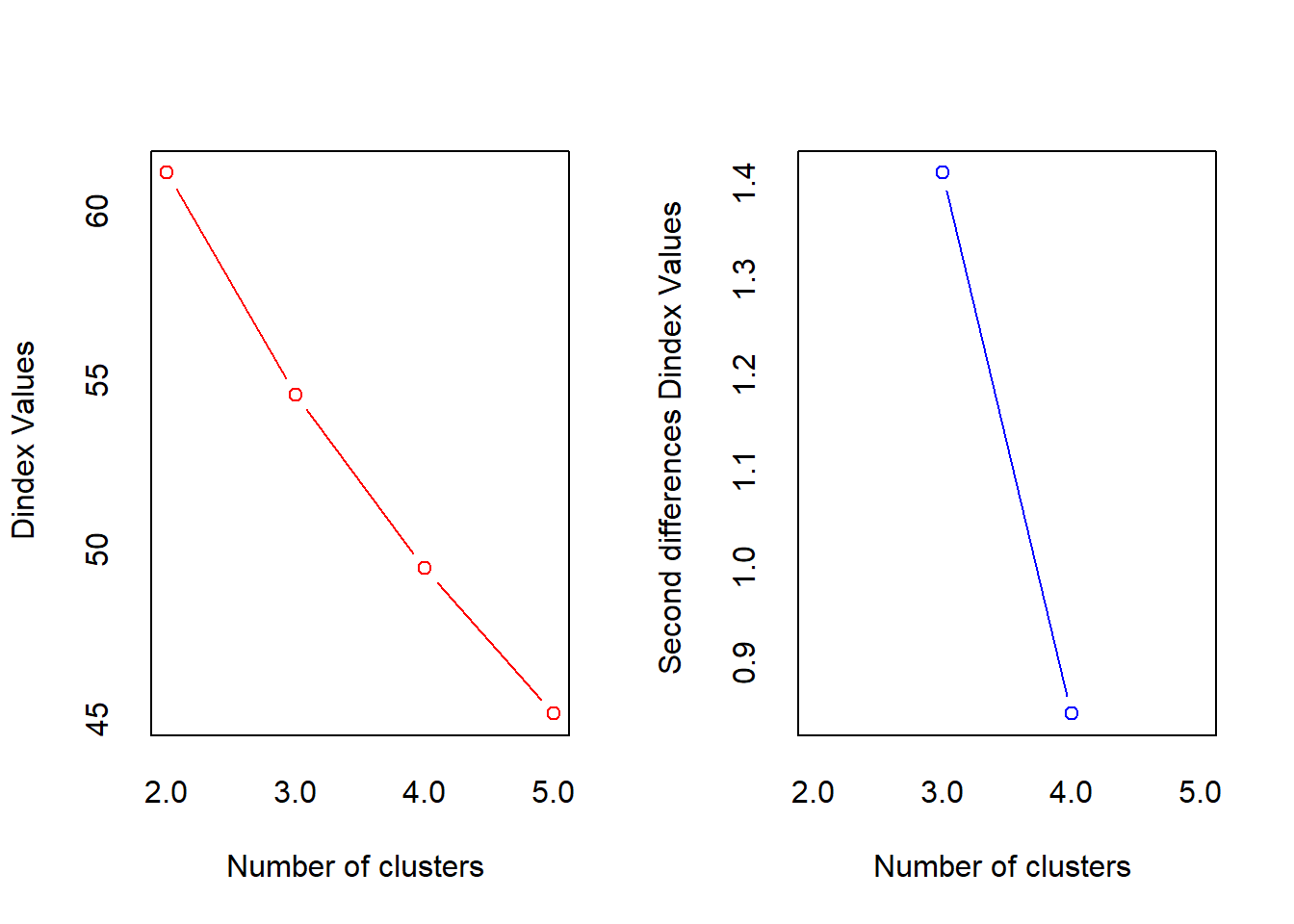
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
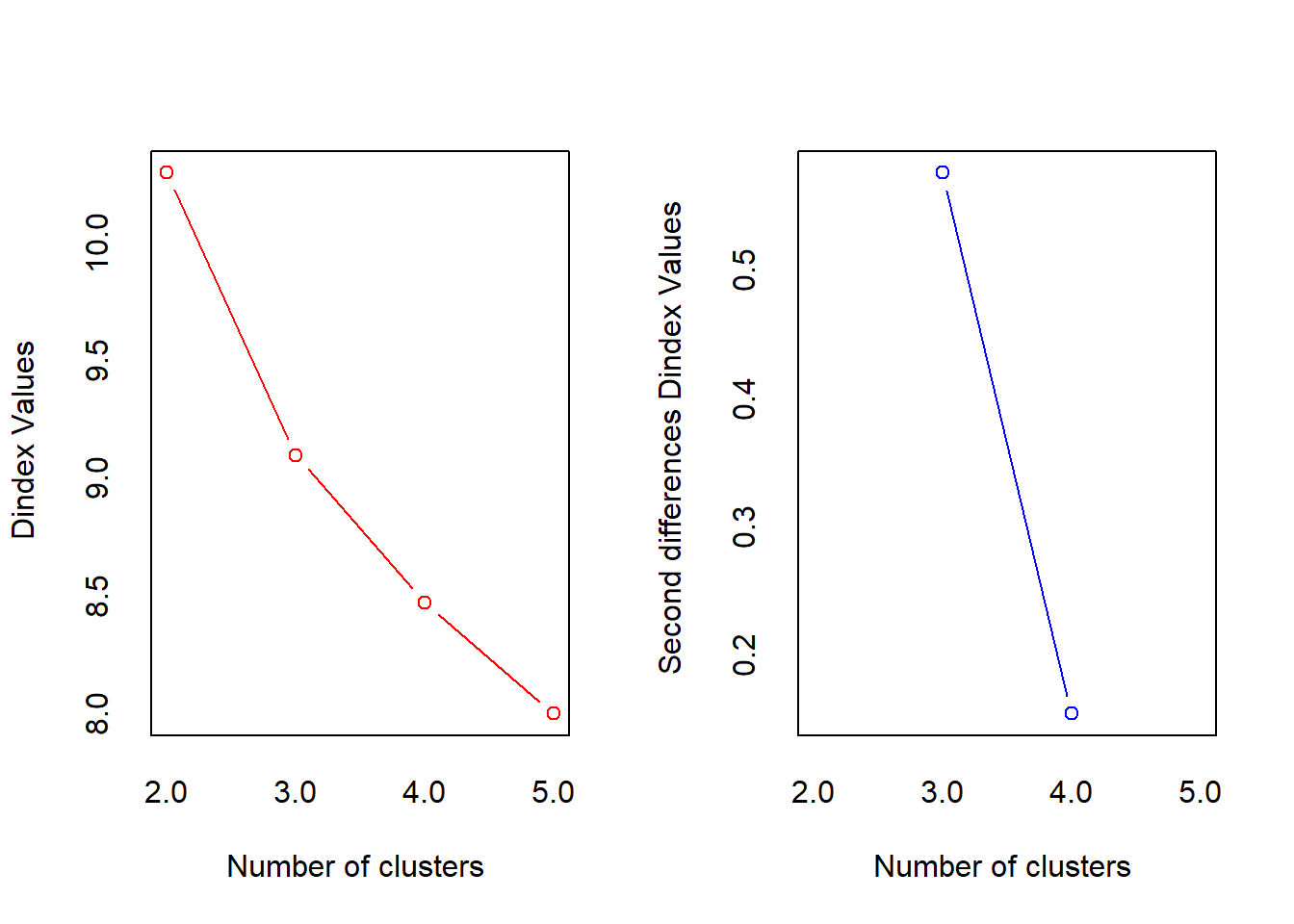
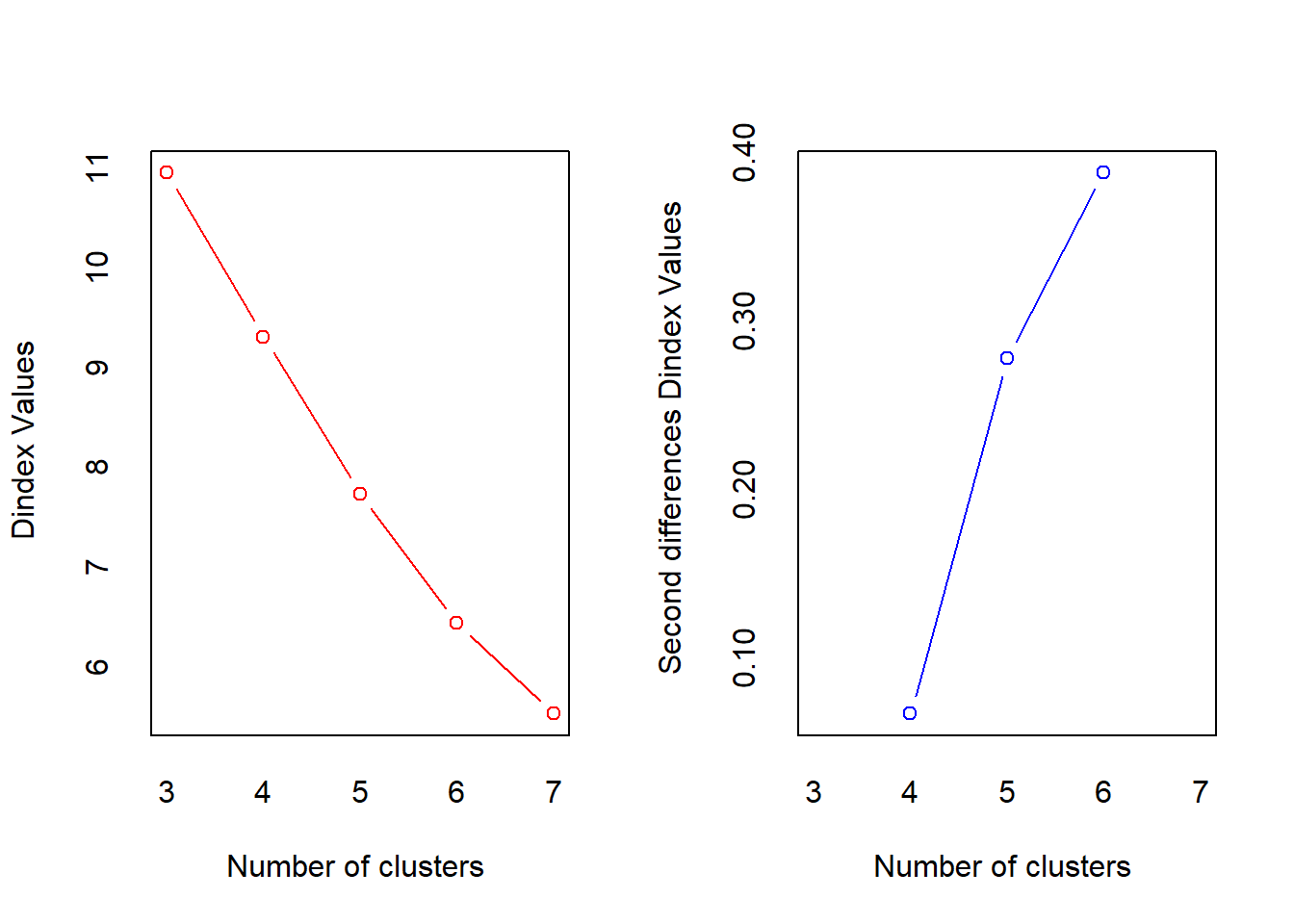
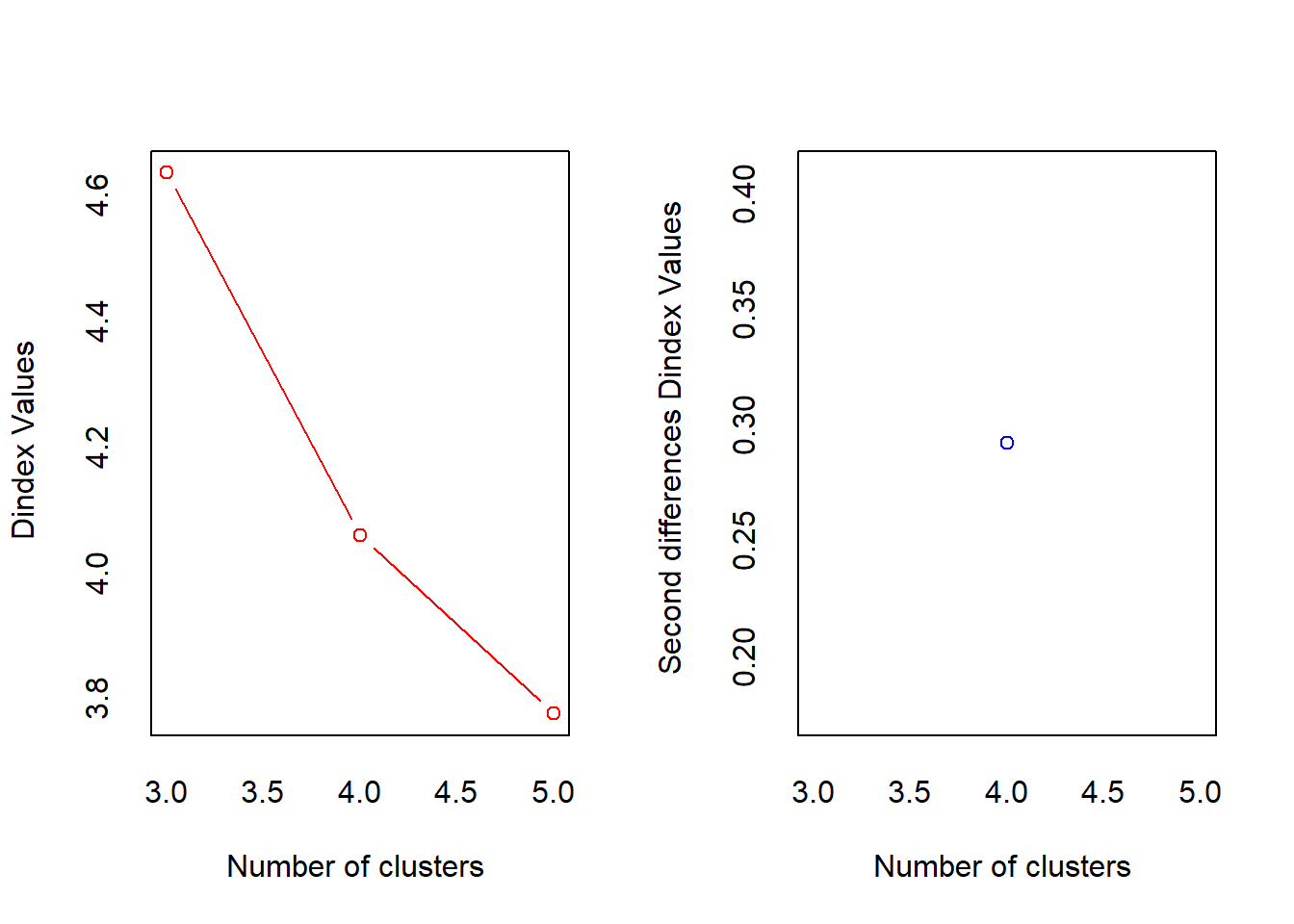
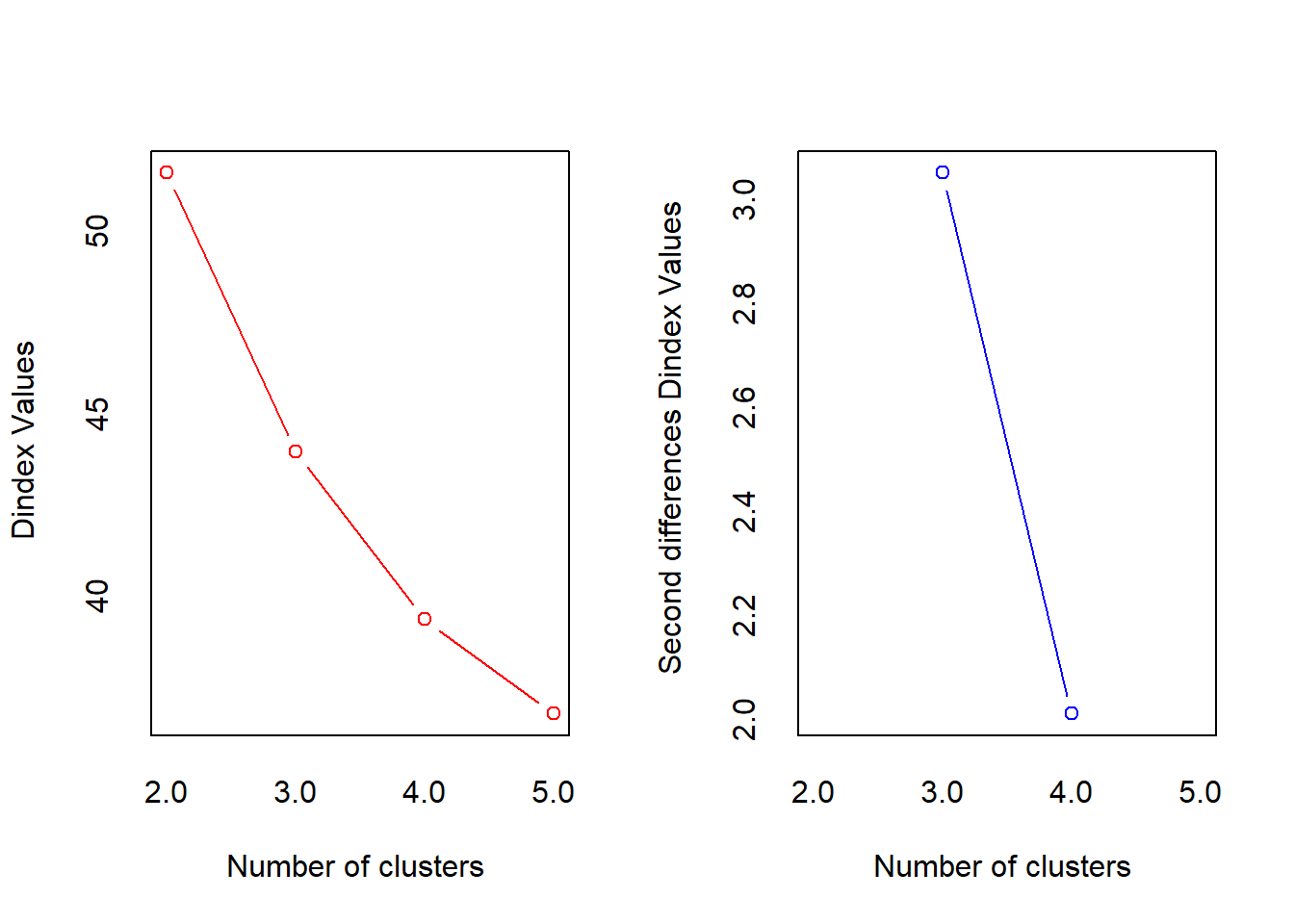
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 10 proposed 2 as the best number of clusters
* 12 proposed 3 as the best number of clusters
* 1 proposed 4 as the best number of clusters
* 1 proposed 5 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 3
*******************************************************************
###
In [30]:
pca.plotr.traj <-function(pca_object, dim.1, dim.2, nb = pca.nb.traj,scale = scaling){ scores <- pca_object$z %>%as_tibble() %>%set_names(paste0('PC', 1:ncol(.))) %>%mutate(State =1:n(), .before =1) %>%mutate(cluster = nb$Best.partition) %>%select(State, cluster, !!sym(dim.1), !!sym(dim.2))# print(scores)# Function to calculate convex hull coordinates for each cluster get_hull_coordinates <-function(cluster_data) { hull_indices <-chull(cluster_data$PC1, cluster_data$PC2) hull_coordinates <- cluster_data[hull_indices, ]return(hull_coordinates) }# Calculate hull coordinates for each cluster renamed_scores <- scores %>% magrittr::set_colnames(c('State', 'cluster', 'PC1', 'PC2')) hull_coordinates <-do.call(rbind, lapply(split(renamed_scores, renamed_scores$cluster), get_hull_coordinates)) %>% magrittr::set_colnames(c('State', 'cluster', dim.1, dim.2))# Calculate the variance explained by each PC var <-signif(pca_object$L/sum(pca_object$L) *100, 2)[c(as.numeric(gsub('PC', '', dim.1)), as.numeric(gsub('PC', '', dim.2)))] representative_states <- renamed_scores %>%left_join( renamed_scores %>%group_by(cluster) %>%summarize(centroid_PC1 =mean(PC1),centroid_PC2 =mean(PC2) ), by ="cluster") %>%mutate(distance_to_centroid =sqrt((PC1 - centroid_PC1)^2+ (PC2 - centroid_PC2)^2) ) %>%group_by(cluster) %>%slice_min(distance_to_centroid) %>%pull(State)# Plot the scores and color by cluster assignment pca_plot <- hull_coordinates %>%ggplot(., aes_string(x = dim.1, y = dim.2)) +geom_polygon(aes(group = cluster, fill =factor(cluster), color =factor(cluster)),alpha =0.05,linewidth =0.75 ) +geom_point(mapping =aes(color =factor(cluster)),data = scores,alpha =0.2 ) +geom_text_repel(data = scores %>%filter(State %in% representative_states),aes(label = State),size =5,fontface ='bold',force =100 ) +geom_point(data = scores %>%filter(State %in% representative_states),aes(fill =factor(cluster)),size =3, shape =21, color ='black', linewidth =0.75 ) +custom.theme(scale) +labs(x = glue::glue(dim.1, ' (', var[1], '%)'),y = glue::glue(dim.2, ' (', var[2], '%)') ) +scale_color_d3(name ='Cluster') +scale_fill_d3(name ='Cluster')return(pca_plot)}
`summarise()` has grouped output by 'resno', 'resid', 'PC'. You can override
using the `.groups` argument.
###
In [32]:
p.pca <- scree +pca.plotr.traj(pca_pdb_traj, 'PC1', 'PC2', nb = pca.nb.traj) + p.loads.traj +plot_layout(design =' AB CC ') &plot_annotation(tag_levels =c('A', 'B', 'C', 'D'))
Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if
`.name_repair` is omitted as of tibble 2.0.0.
ℹ Using compatibility `.name_repair`.
Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
ℹ Please use tidy evaluation idioms with `aes()`.
ℹ See also `vignette("ggplot2-in-packages")` for more information.
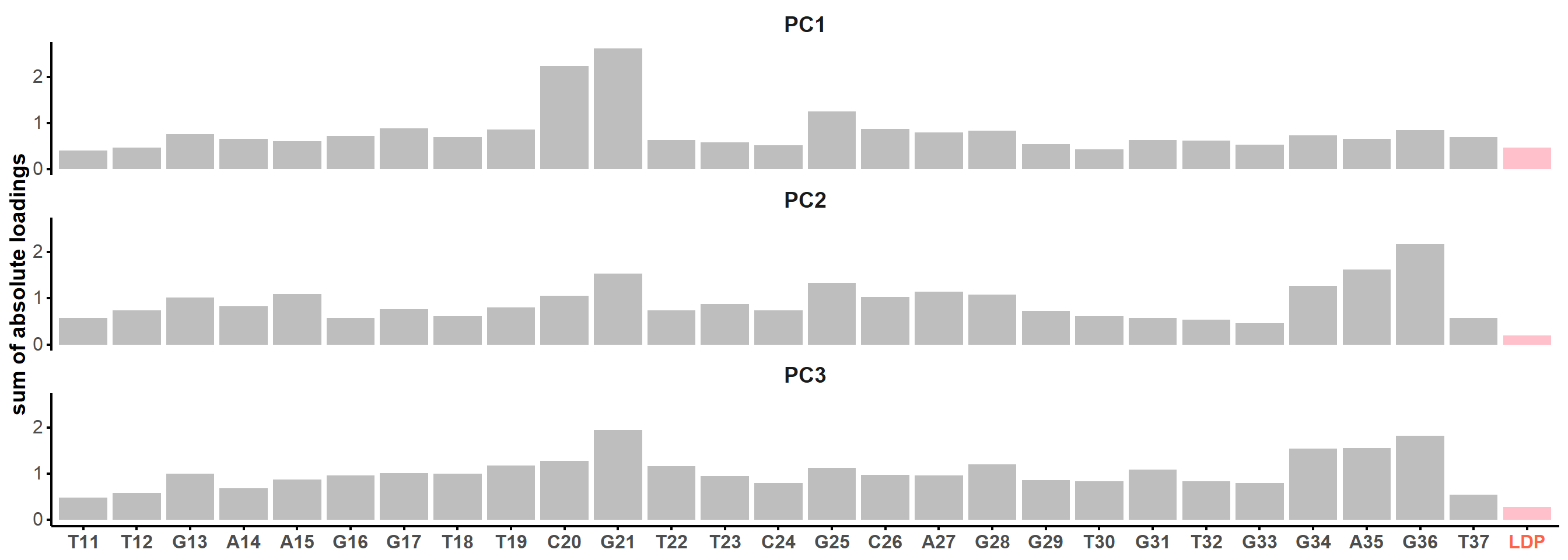
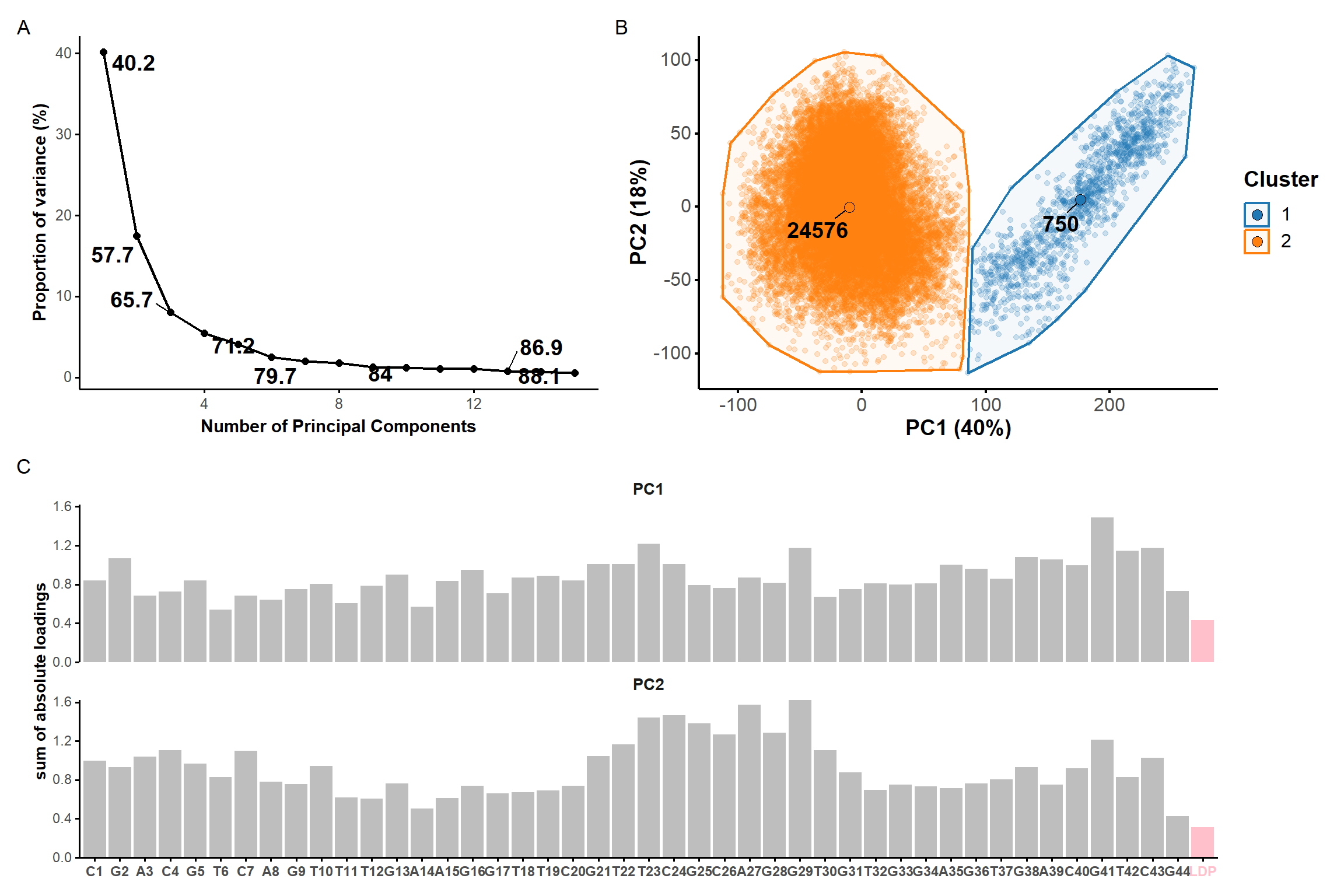
Figure 7: Principal component analysis on the coordinates of the restrained microsecond trajectory. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points. (B) Score plots along the first two principal components, colored by kmeans clusters. (C) Sum of absolute loadings of residues for the first two first principal components
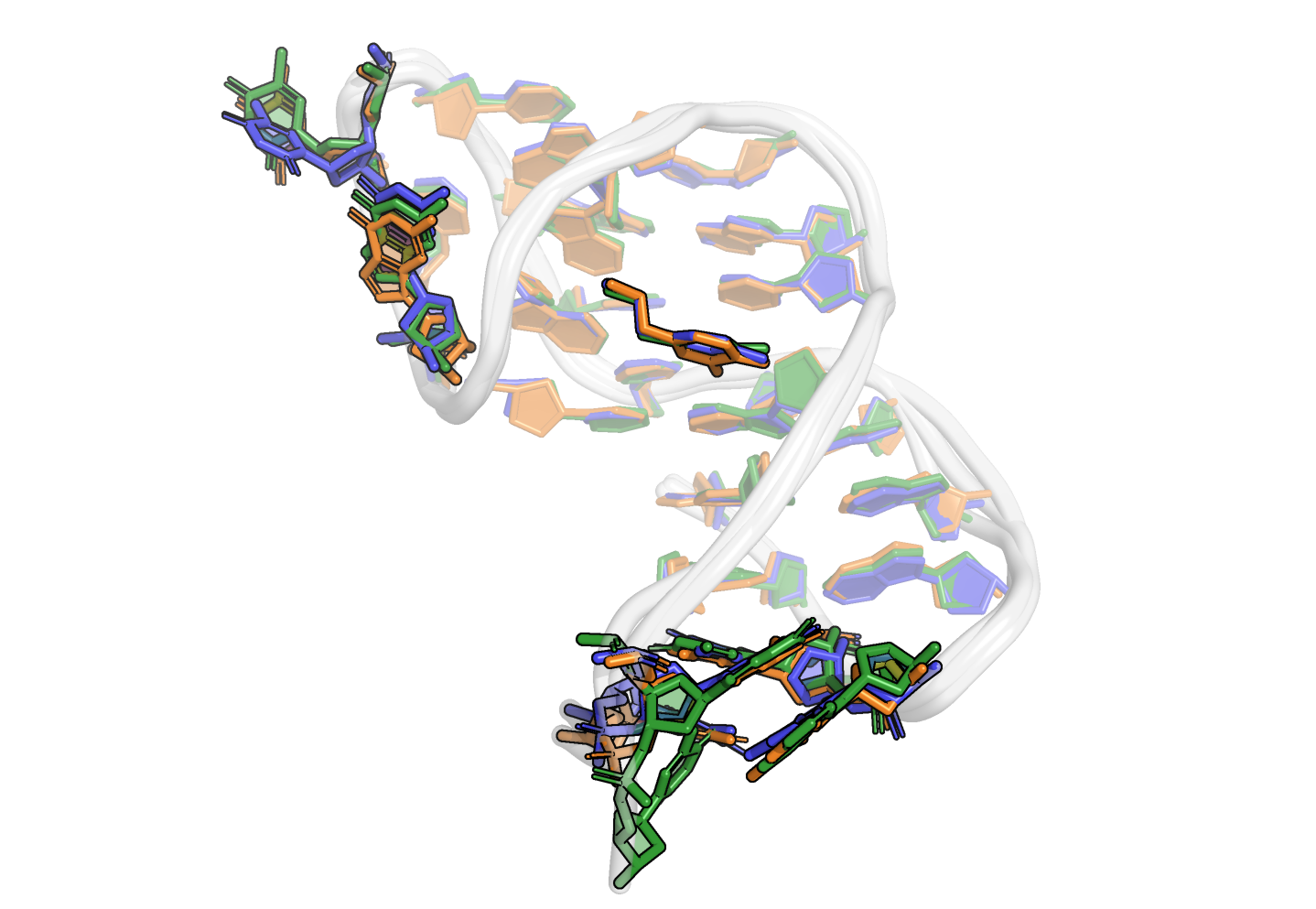
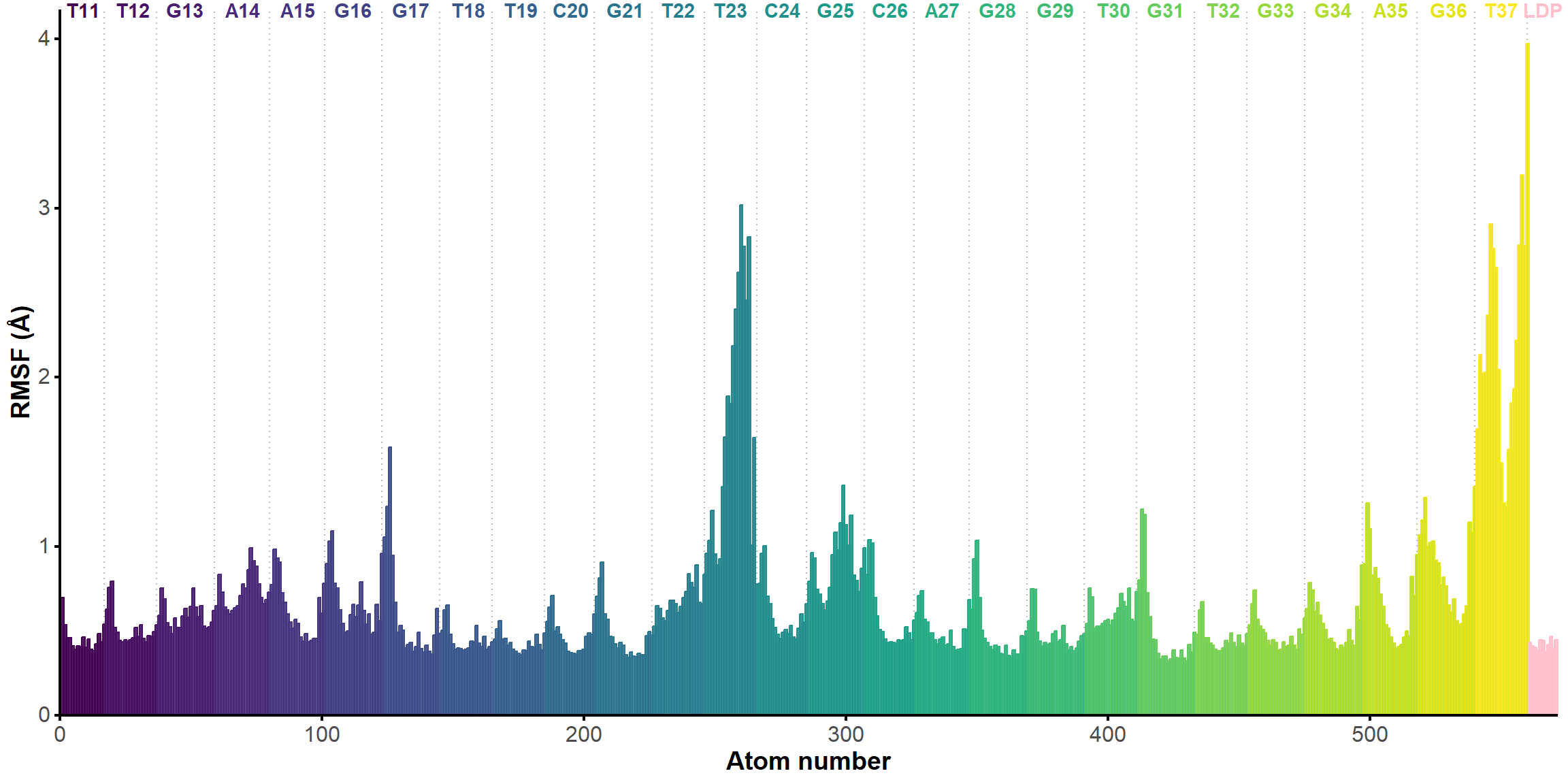
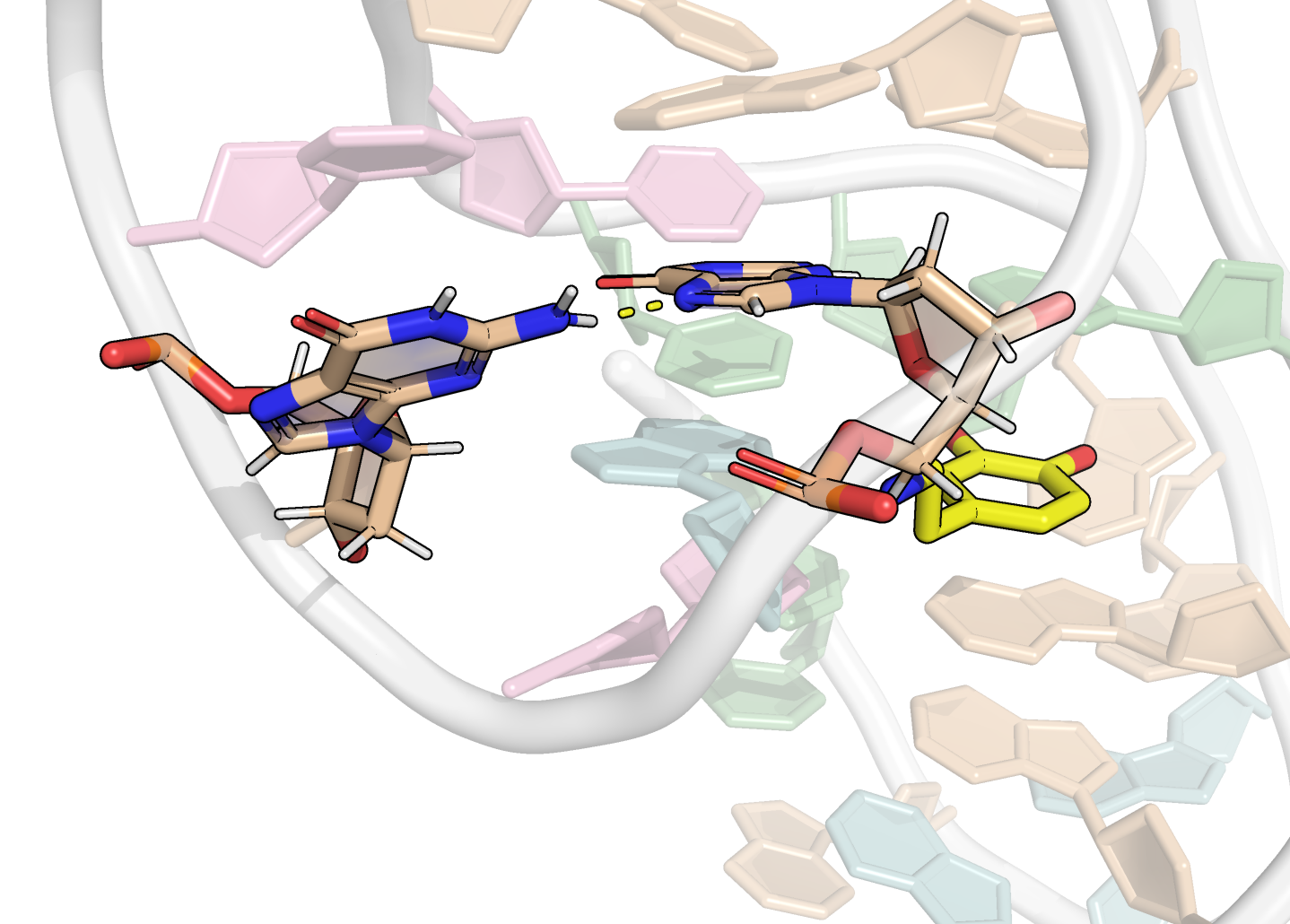
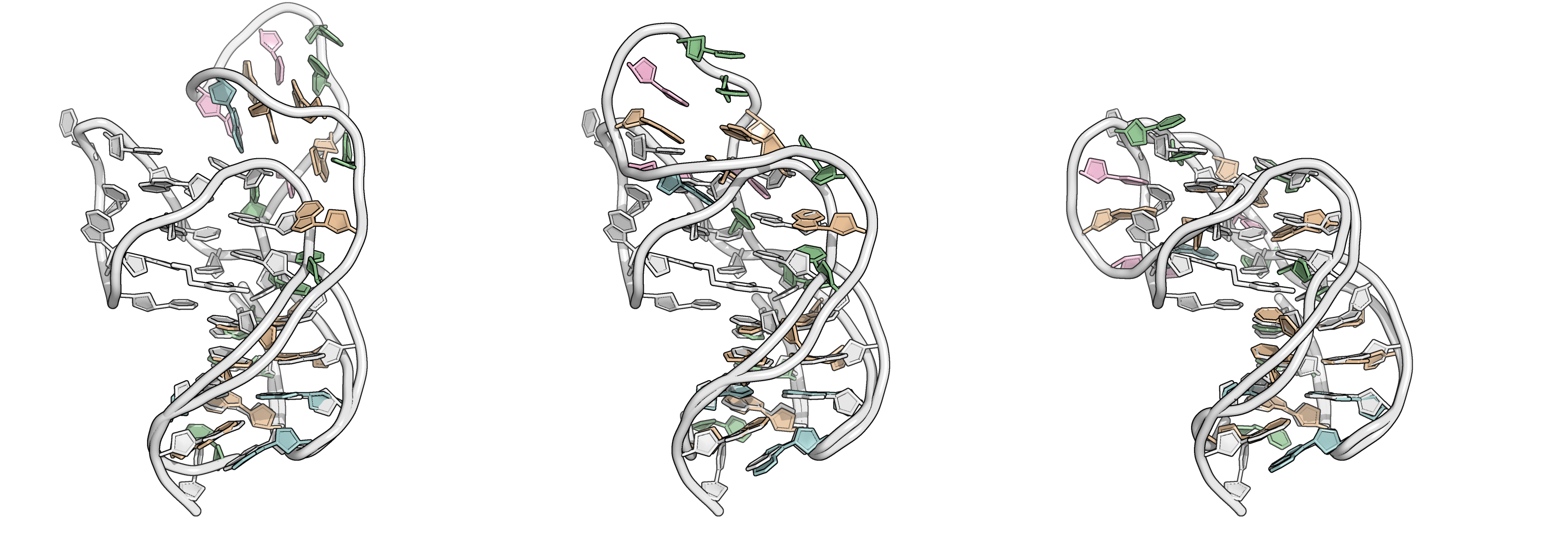
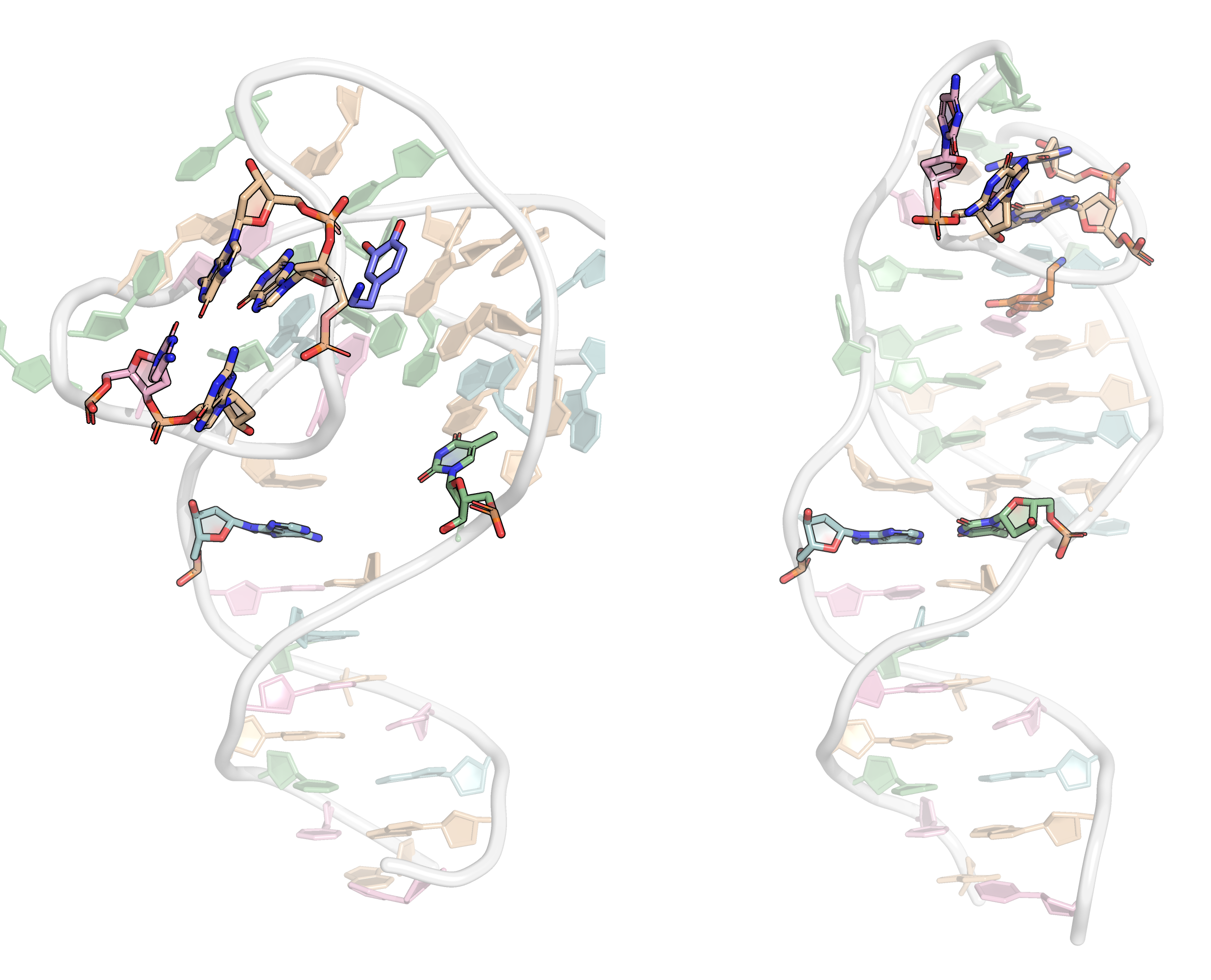
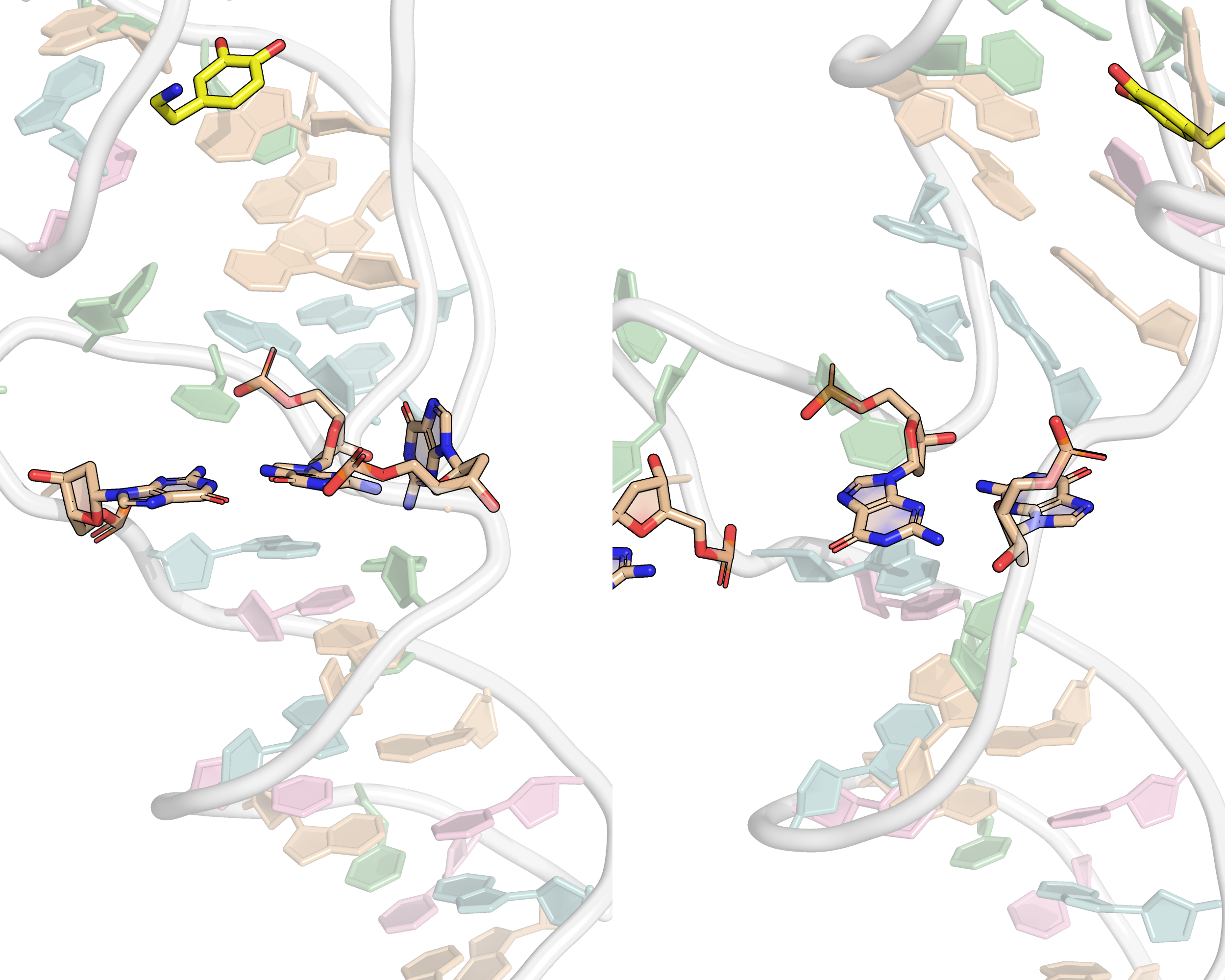
From the first four principal components, the data is best clustered in three groups. The states closest to the centroids of each cluster are shown below. The largest differences are at the 3’-end region (G36:T37 and G13:A14, different in cluster 3), the T23 chain reversal loop (different in cluster 1), and - to a lower extent - the G25. This correlates well with the root mean squared flucturations measured during the simulation (Figure 8)
Representative structures of the two PCA clusters. Differences are highlighted.
Warning: `position_stack()` requires non-overlapping x intervals
`position_stack()` requires non-overlapping x intervals
###
Figure 8: Root mean squared fluctuations (RMSF), highlighting the residues with the largest mobility during the restrained simulation.
2.1.5 Cross-correlation analysis
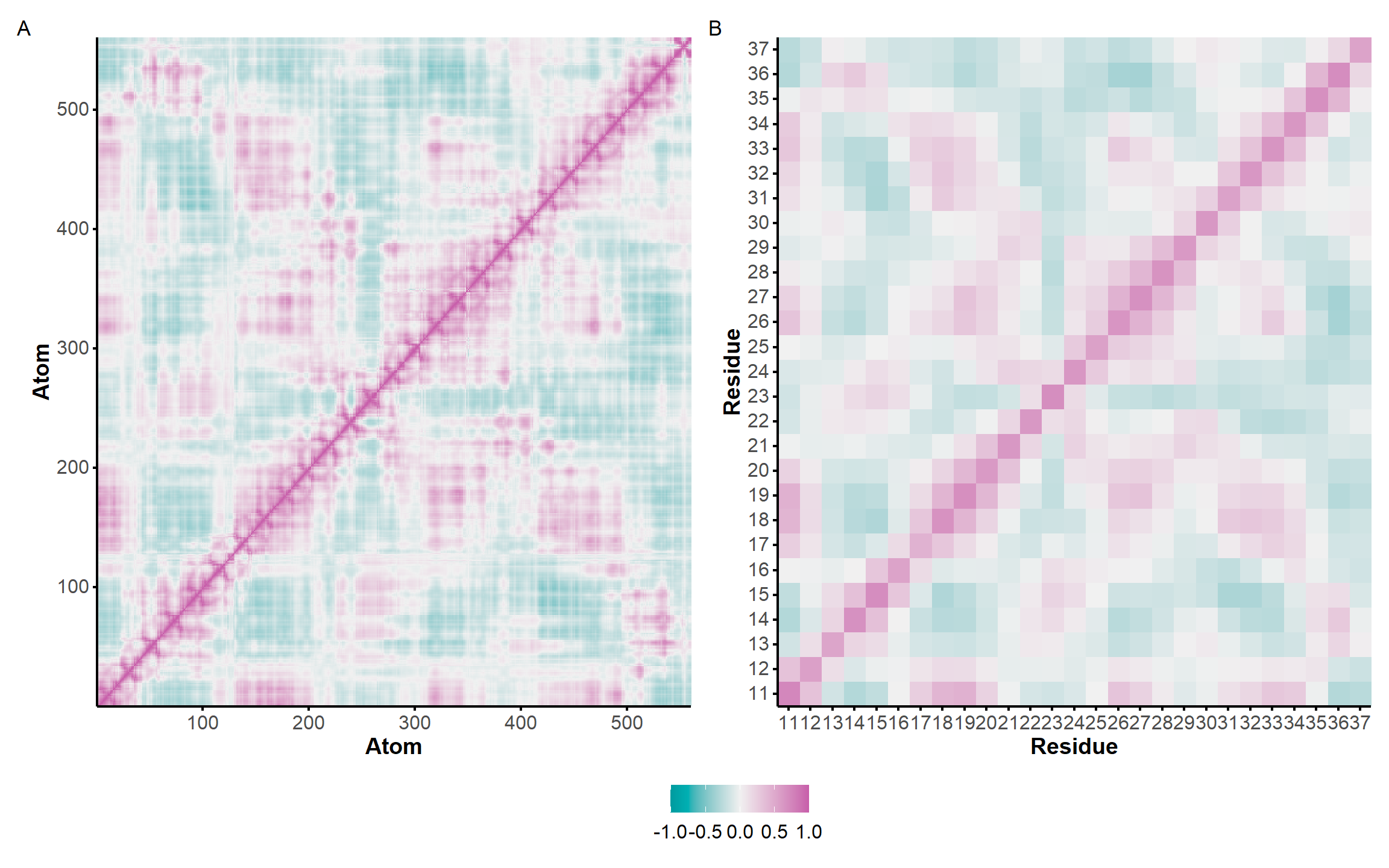
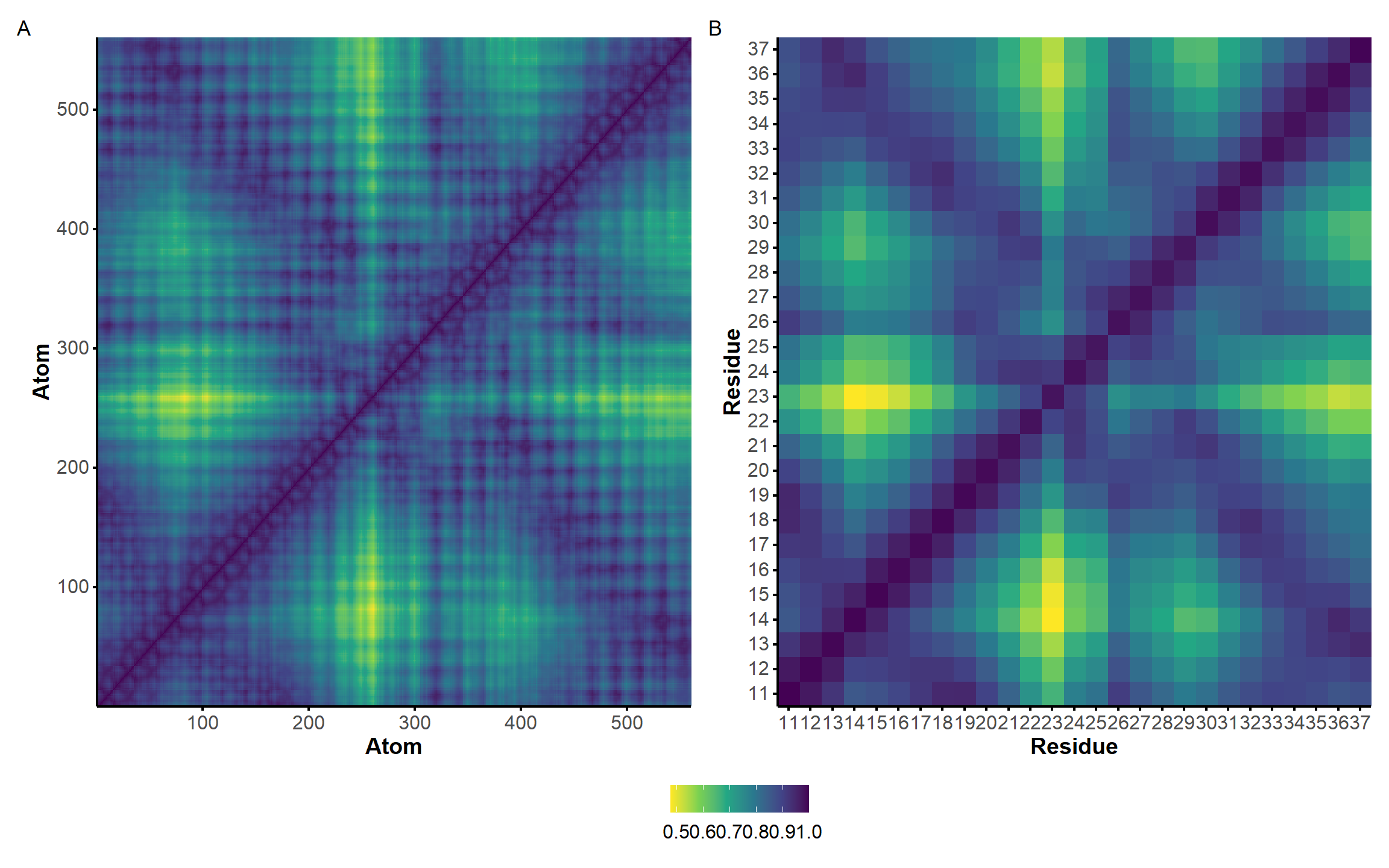
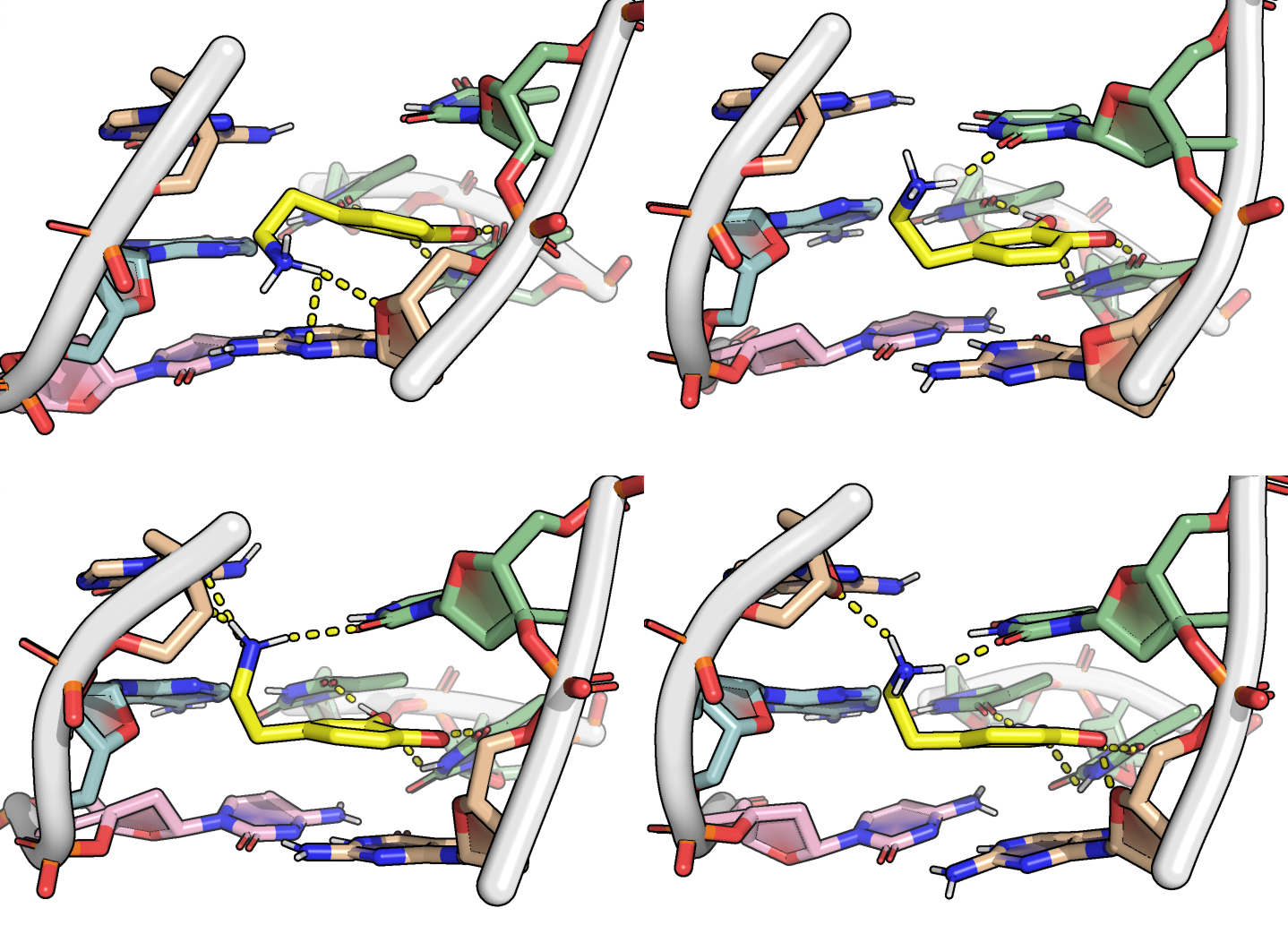
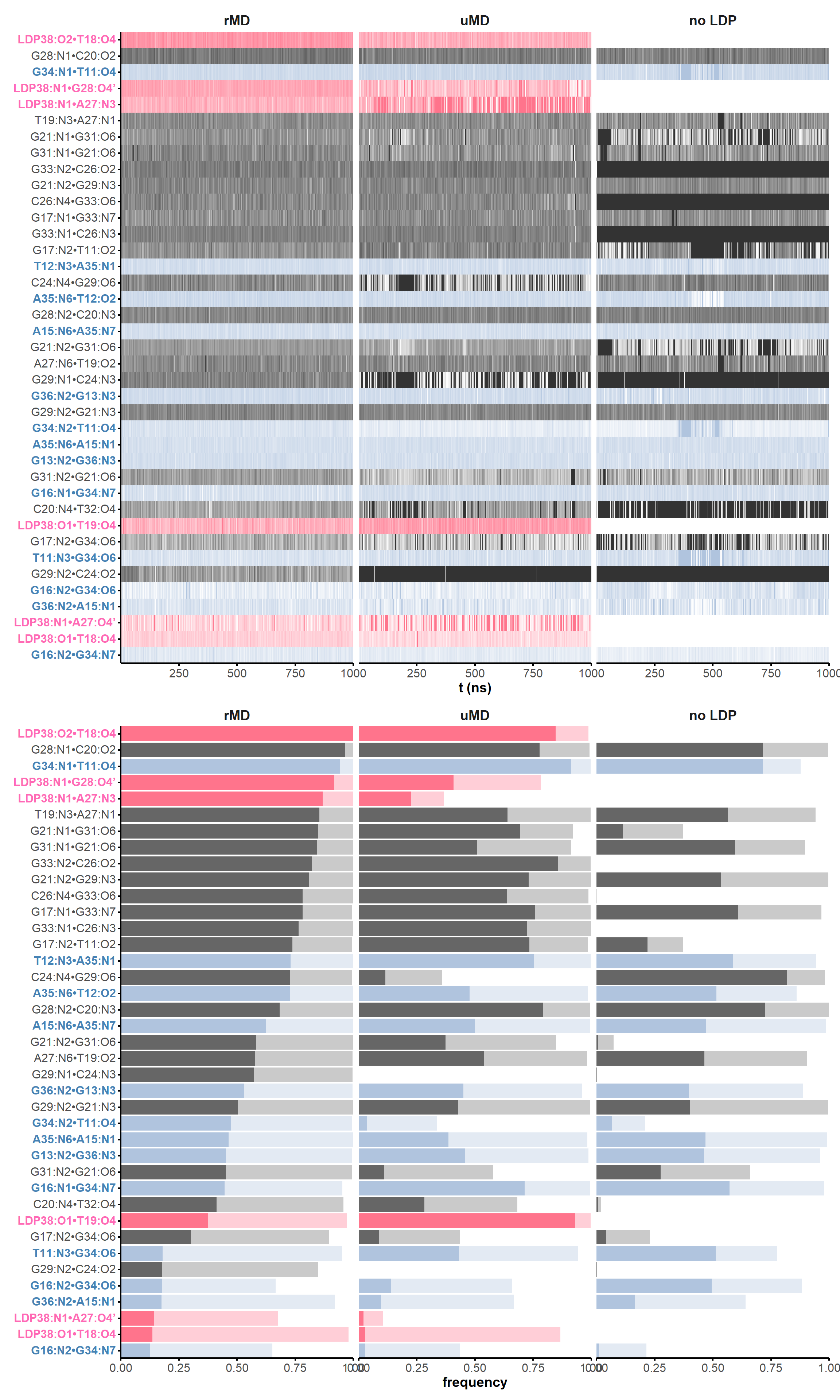
The correlation of atomic movements was assessed by calculation of the atom-wise cross-correlation matrix, shown in Figure 9 as a heatmap. Negative values indicate that the atoms/residues move in opposite directions.
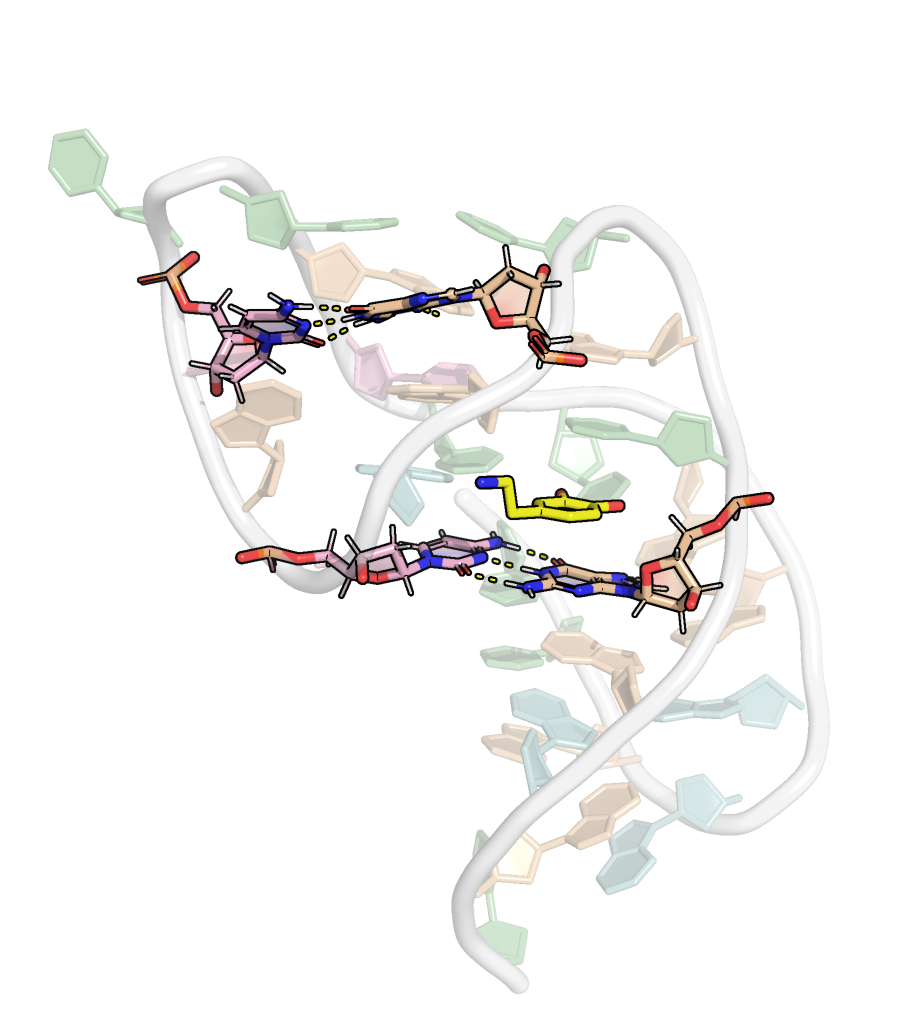
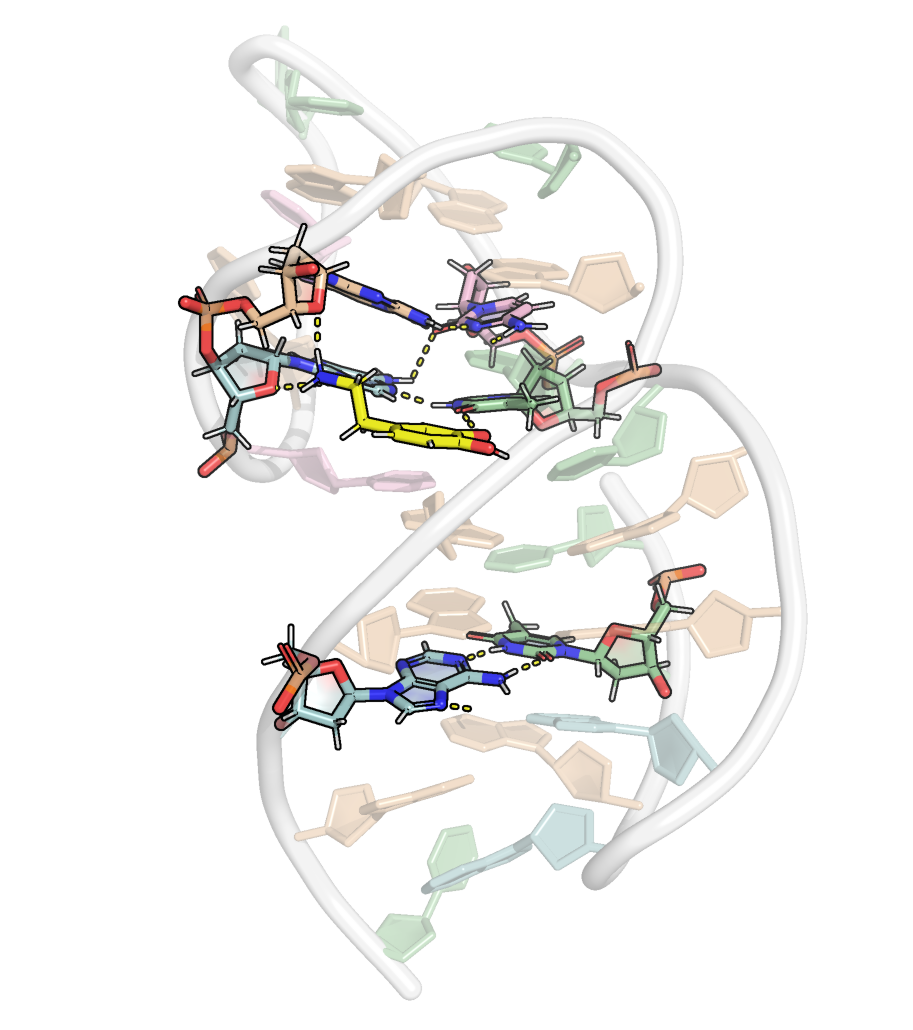
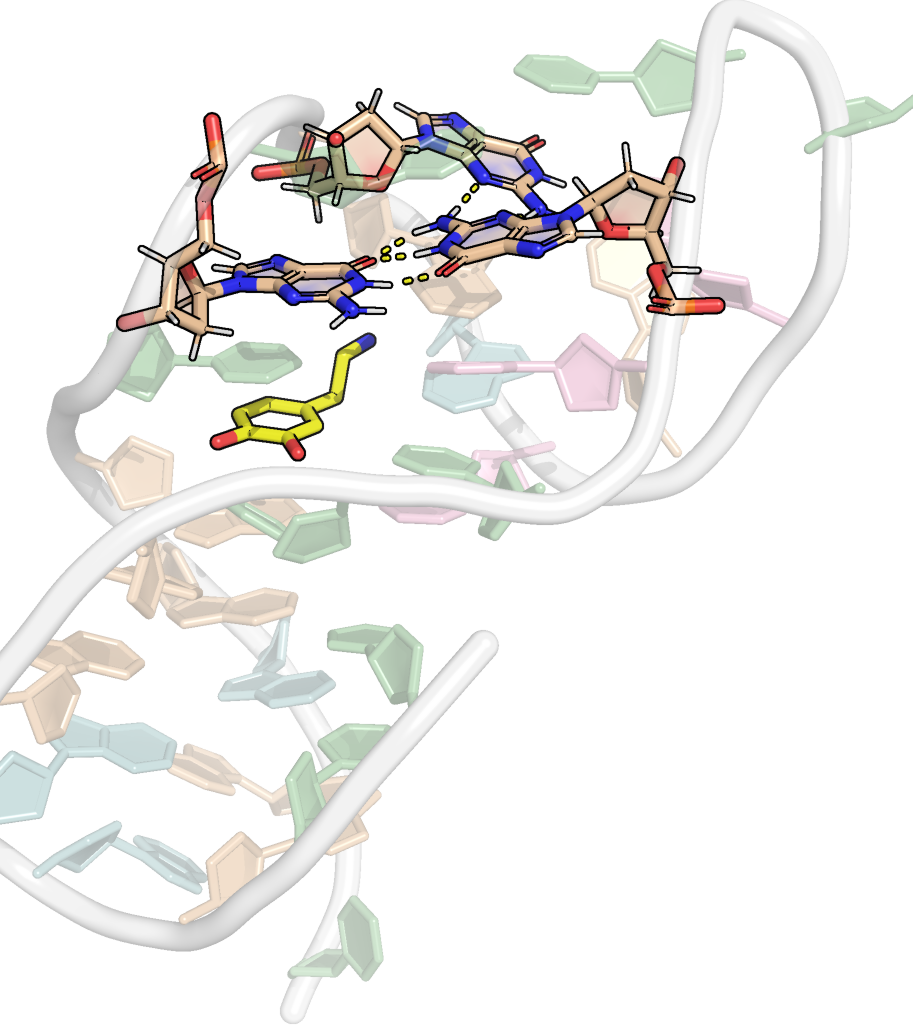
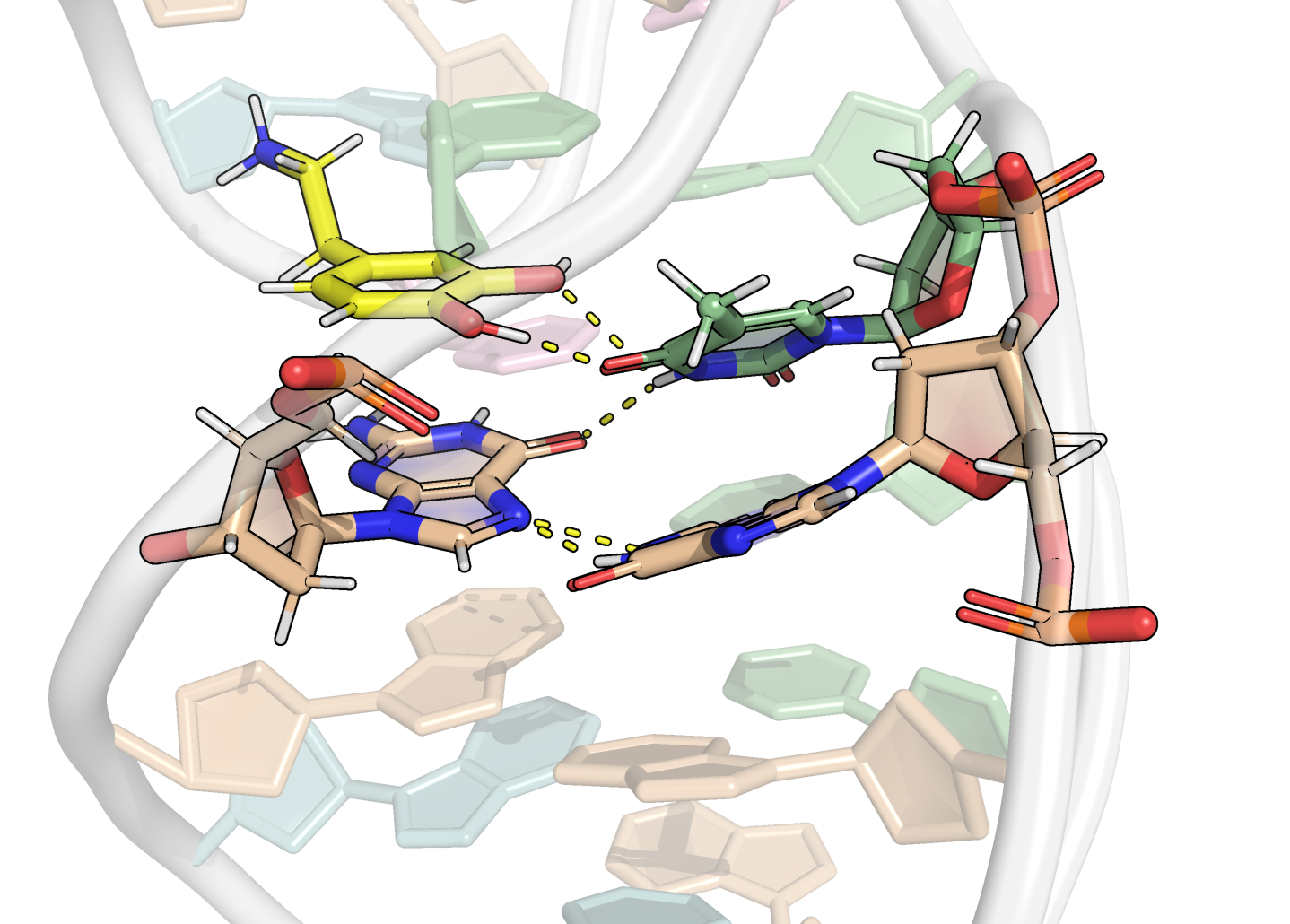
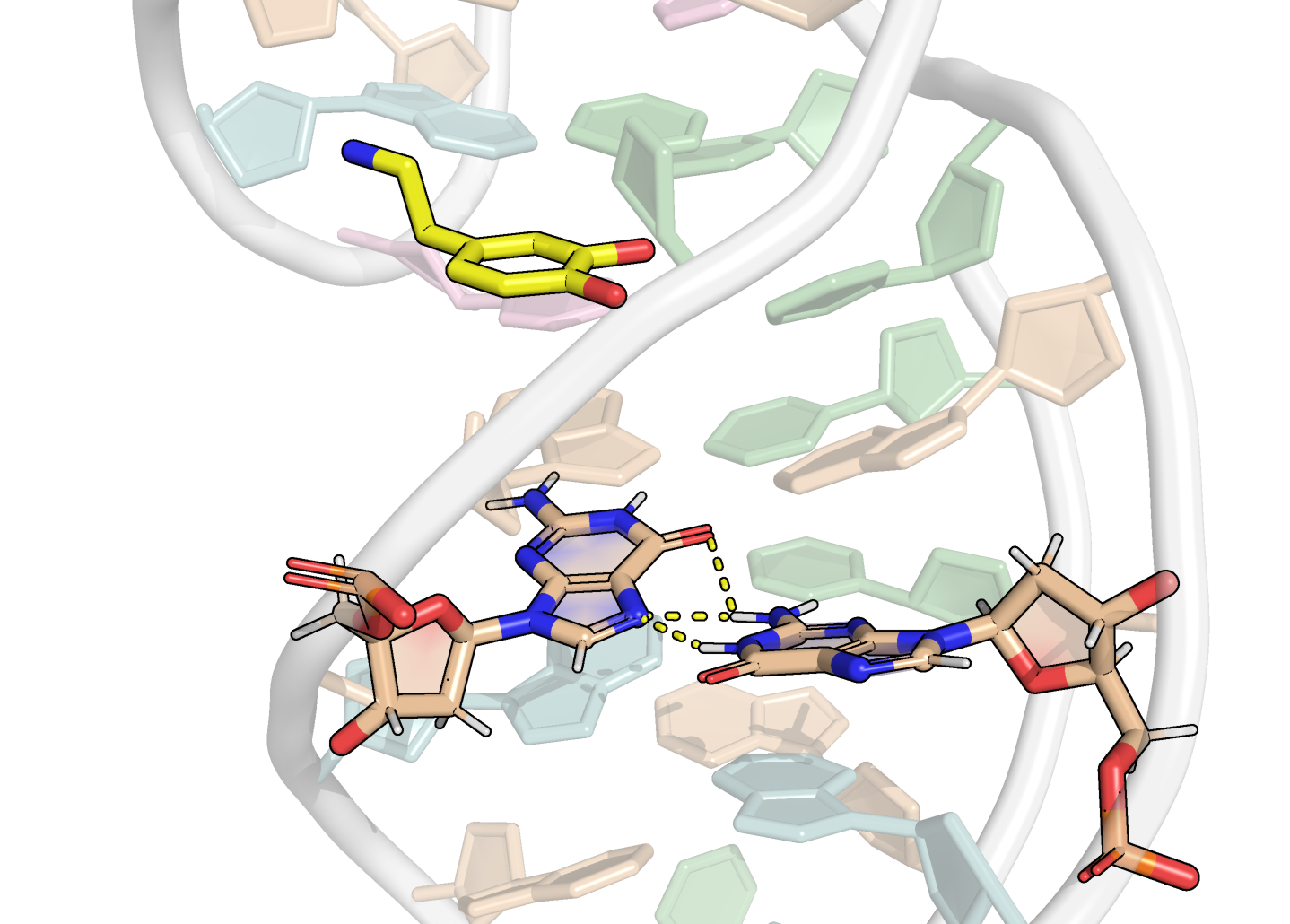
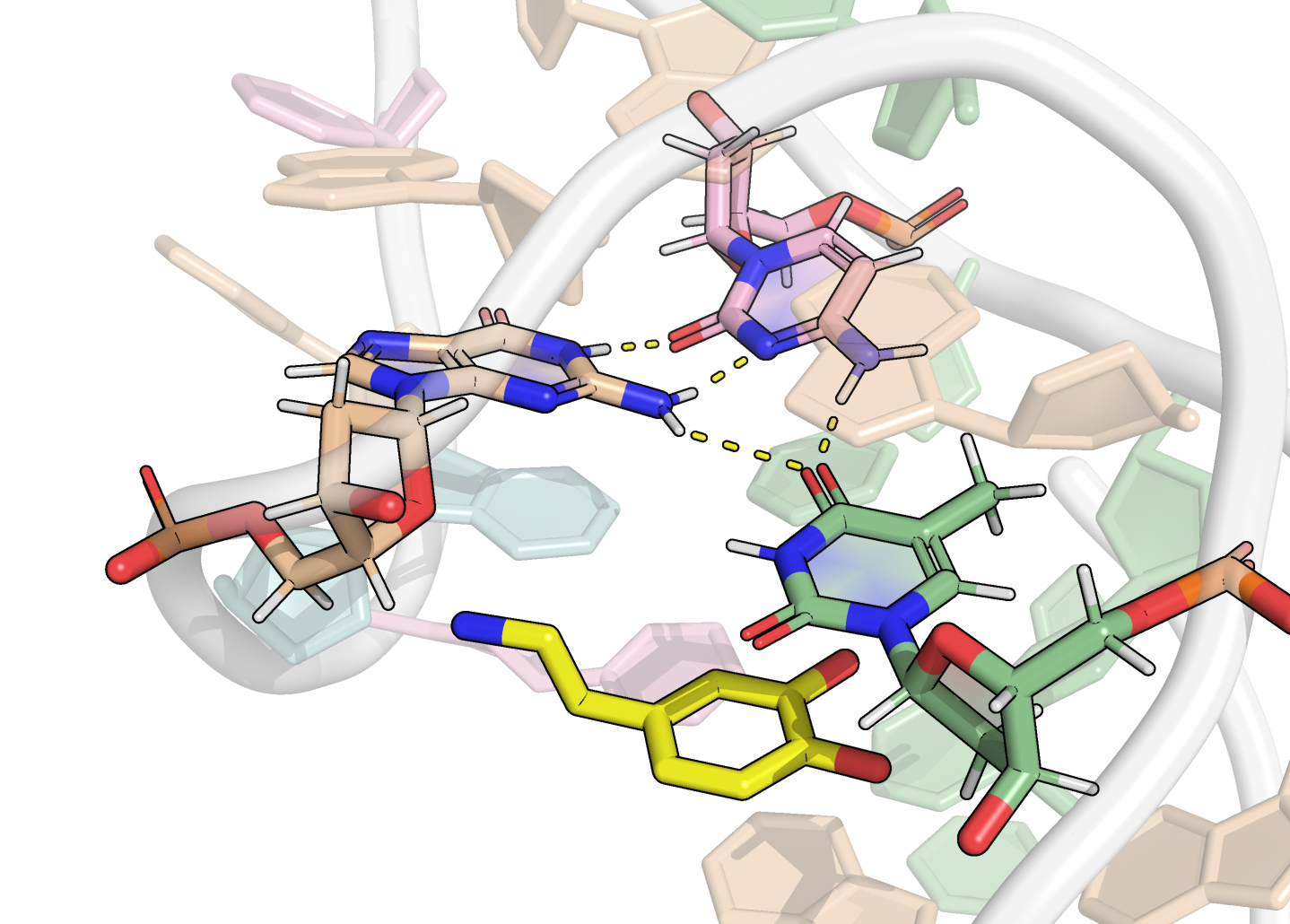
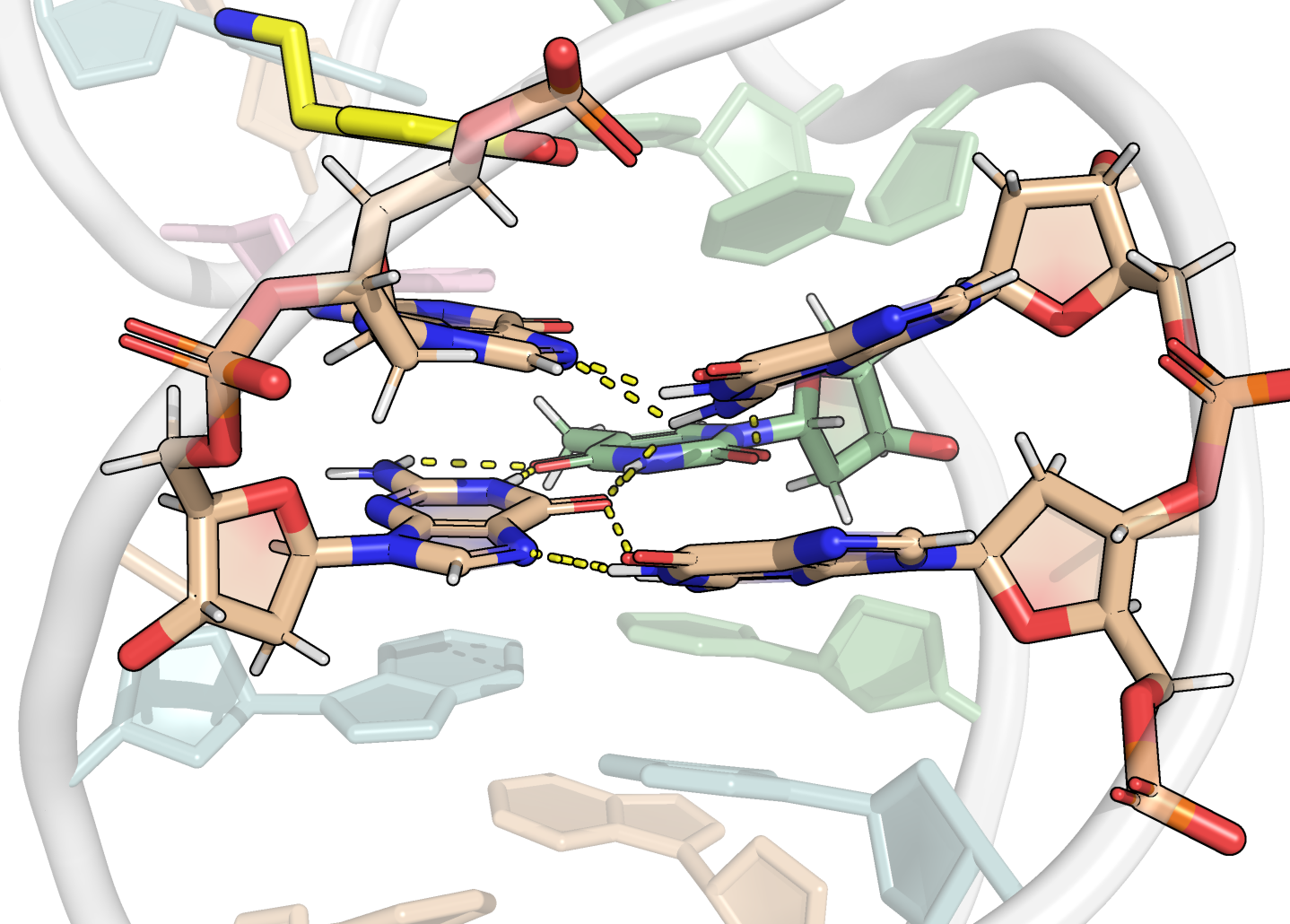
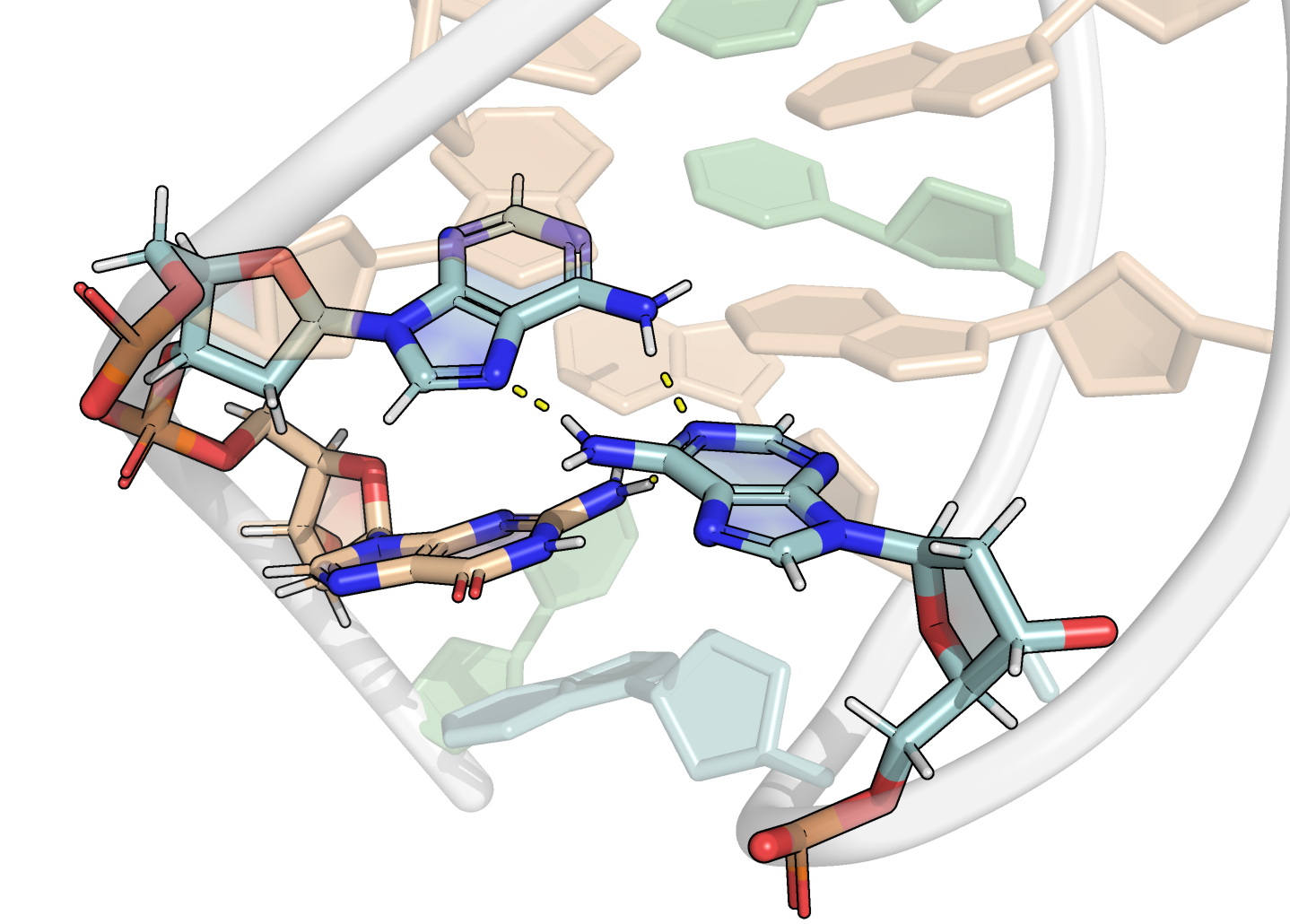
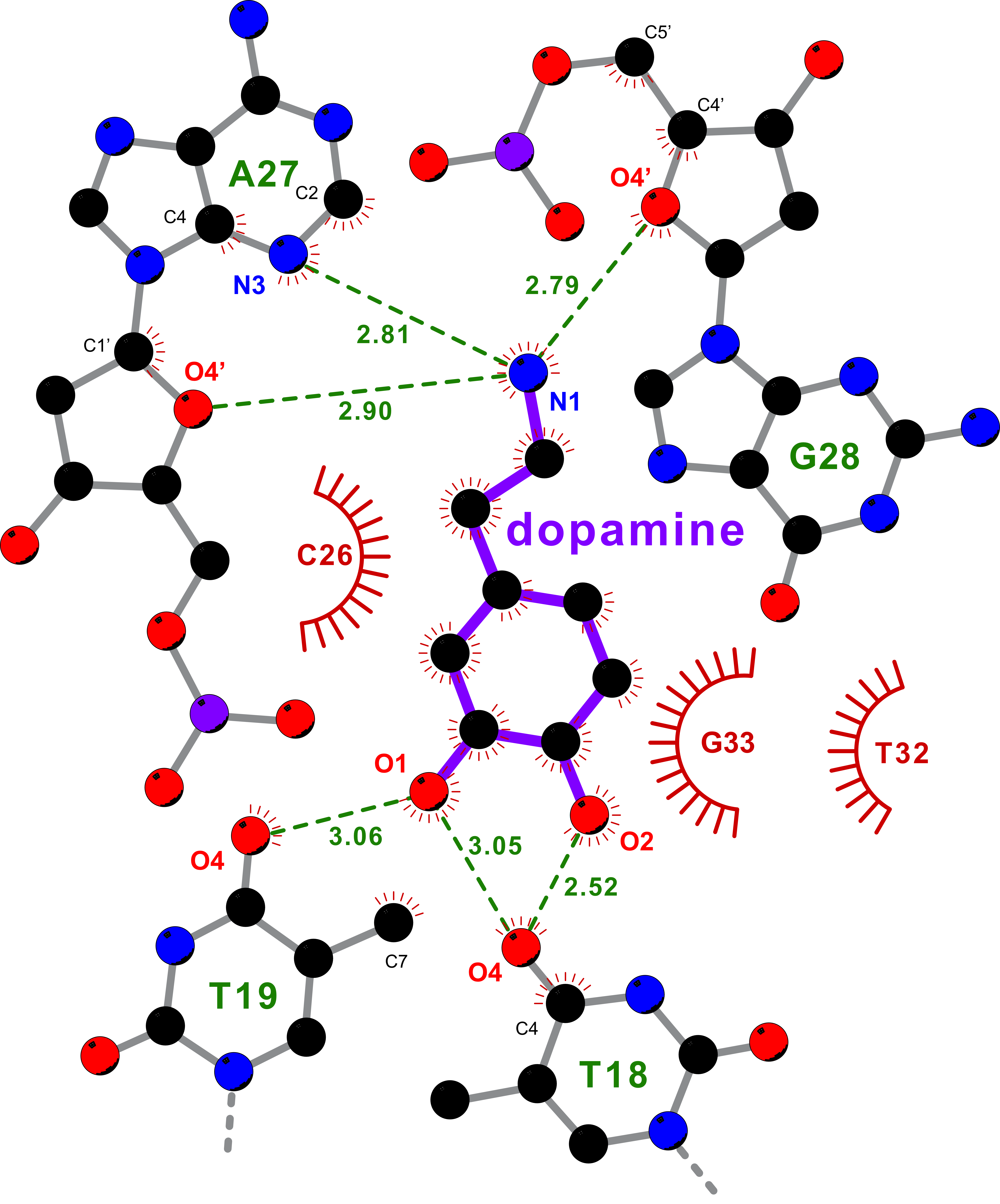
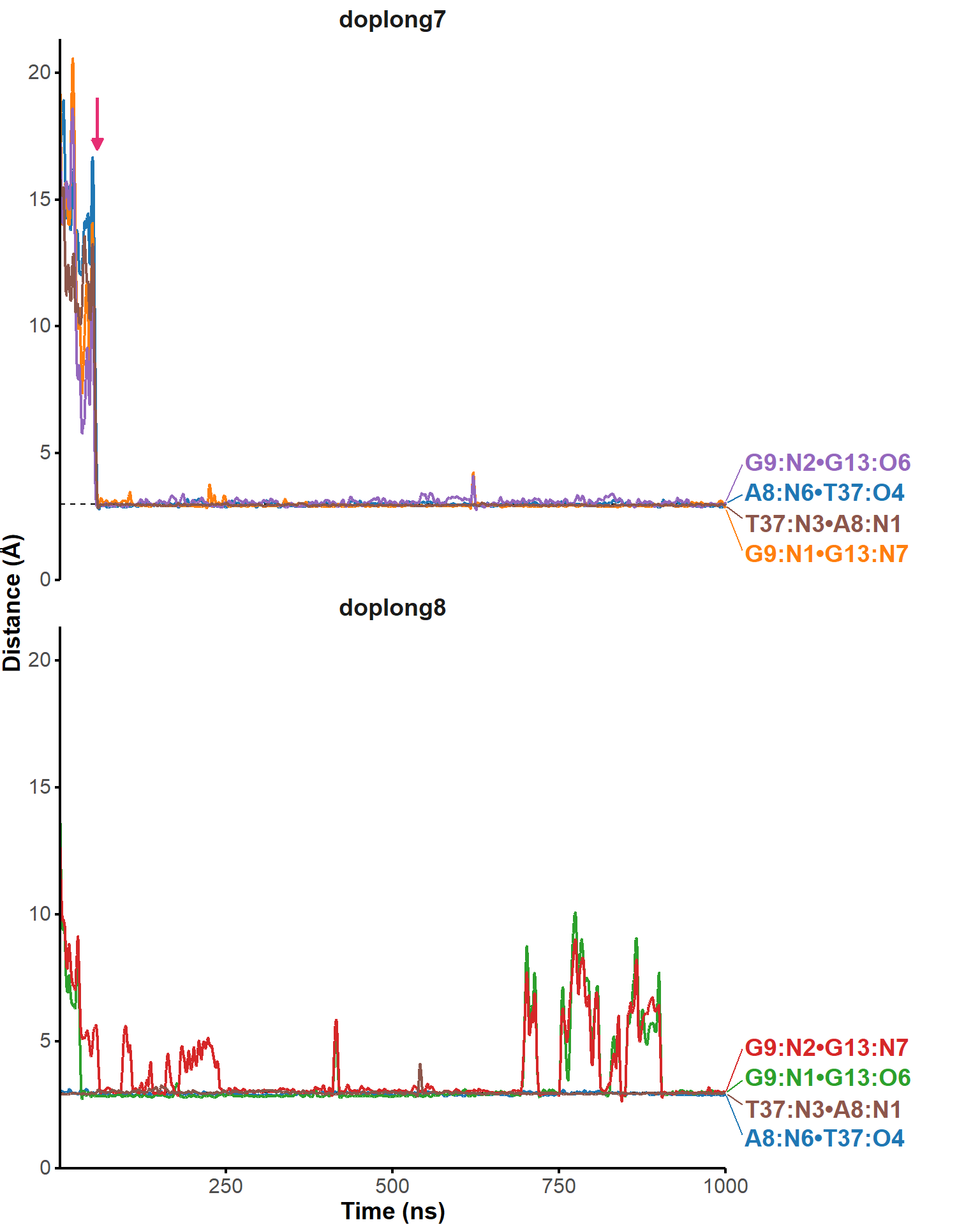
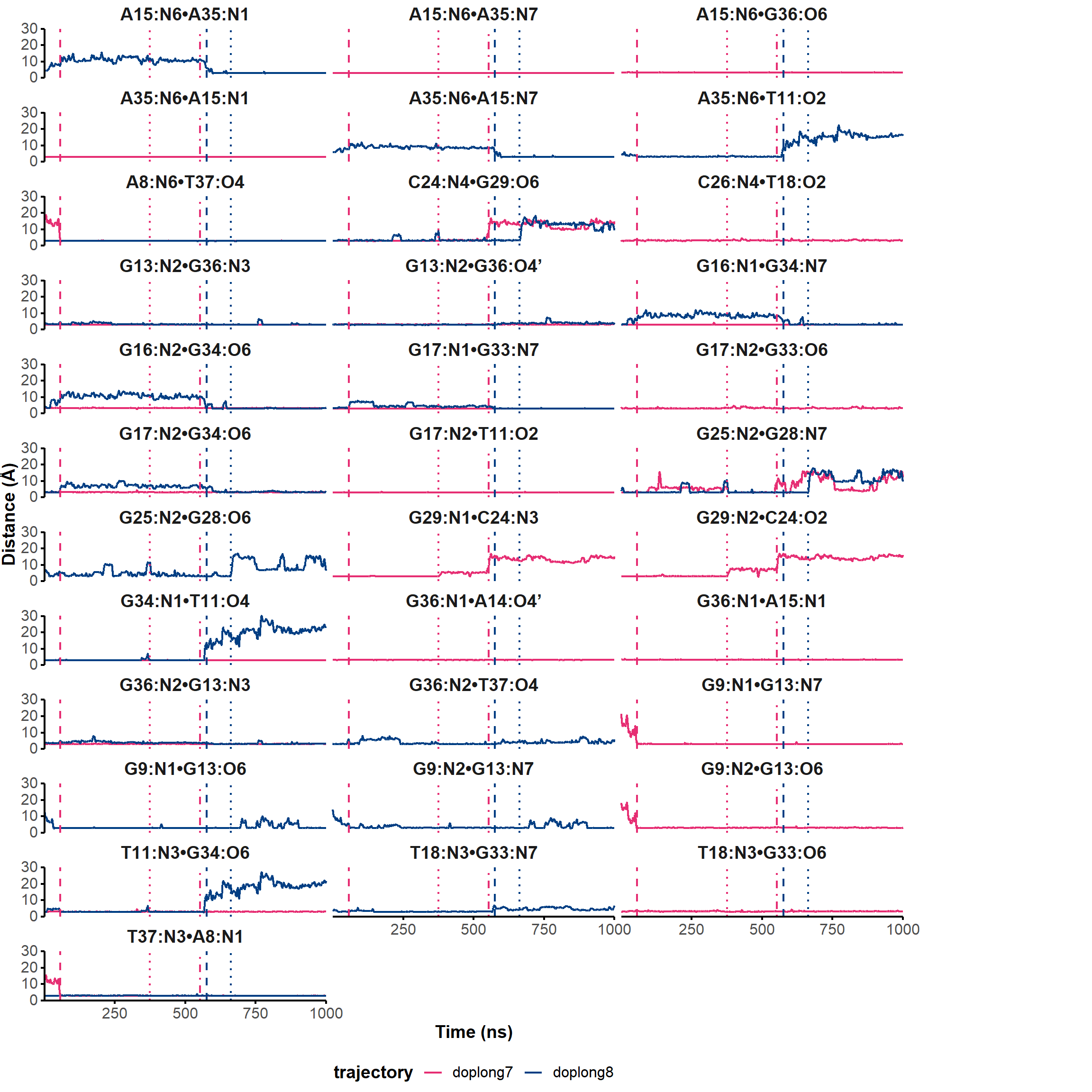
Here, it is possible to observe a positive correlation of the dopamine binding site residues T18 and T19 H-bonded to the cathecol, A27 and G28 to the ammonium, and the stacked C26, T32 and G33 (see Section 2.5 below).
Figure 9: Dynamical cross-correlation map (DCCM) of the microsecond simulation across atoms (A) or residues (B)
2.1.6 Domain analysis
The identification of geometrically rigid regions of the aptamer in the microsecond simulation was performed with GeoStaS algorithm (Romanowska, Nowiński, and Trylska 2012). In Figure 10, we see that most of the aptamer is rigid, with the exception of the T23 loop and, to a lower extent, the nearby T30 loop.
In [37]:
gs <-geostas( pdb.long$xyz[seq(1, nrow(pdb.long$xyz), by =nrow(pdb.long$xyz)/1000),], fit =FALSE)
.. 'xyz' coordinate data with 1000 frames
.. coordinates are not superimposed prior to geostas calculation
.. calculating atomic movement similarity matrix ('amsm.xyz()')
.. dimensions of AMSM are 571x571
.. clustering AMSM using 'kmeans'
Figure 10: Atomic movement similarity matrix obtained with the GeoStaS analysis for the microsecond simulation across atoms (A) or residues (B). The calculation was carried out on 1000 evenly-spaced frames.
2.2 Minimized structures
In [39]:
rmsd.interstates <-rmsd(pdb, a.inds =atom.select(pdb),b.inds =atom.select(pdb),fit =TRUE) %>%as.data.frame() %>% magrittr::set_colnames(as.character(1:15)) %>%mutate(State =1:15, .before =1) %>%#replace all 0 with NA in columns 2:16mutate(across(2:16, ~ifelse(. ==0, NA_real_, .))) %>%rbind(c(NA_real_, sapply(.[, -1], mean, na.rm =TRUE))) %>%mutate(State =ifelse(is.na(State), 'Mean', as.character(State)))# Mean RMSDmean.rmsd.doprstslt <- rmsd.interstates[rmsd.interstates$State =="Mean", 2:16] %>%#collapse into a single vectorunlist() %>%mean(.) %>%round(1)# Std. dev. of RMSDsd.rmsd.doprstslt <- rmsd.interstates[rmsd.interstates$State =="Mean", 2:16] %>%#collapse into a single vectorunlist() %>%sd(.) %>%round(2)
2.2.1 Structures and energies
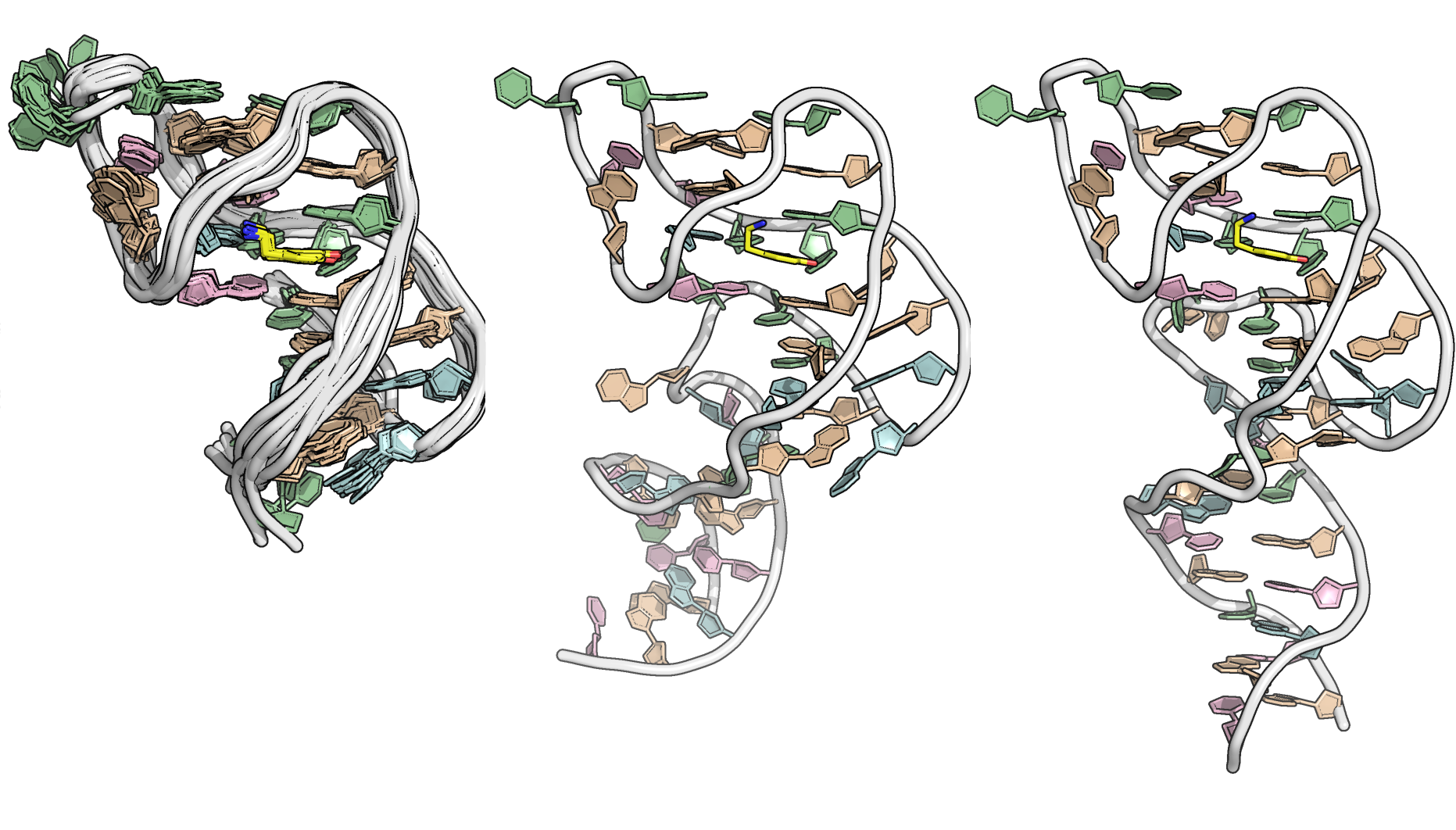
The fifteen minimized structures of the dopamine aptamer obtained by restrained molecular dynamics are displayed in Figure 11 (mean RMSD across structures: 1.3 \(\pm\) 0.14 Å). The longer sequence (with duplex stem at the bottom) are shown alongside (also minimized after 10 ns rMD ; see Section 2.7).
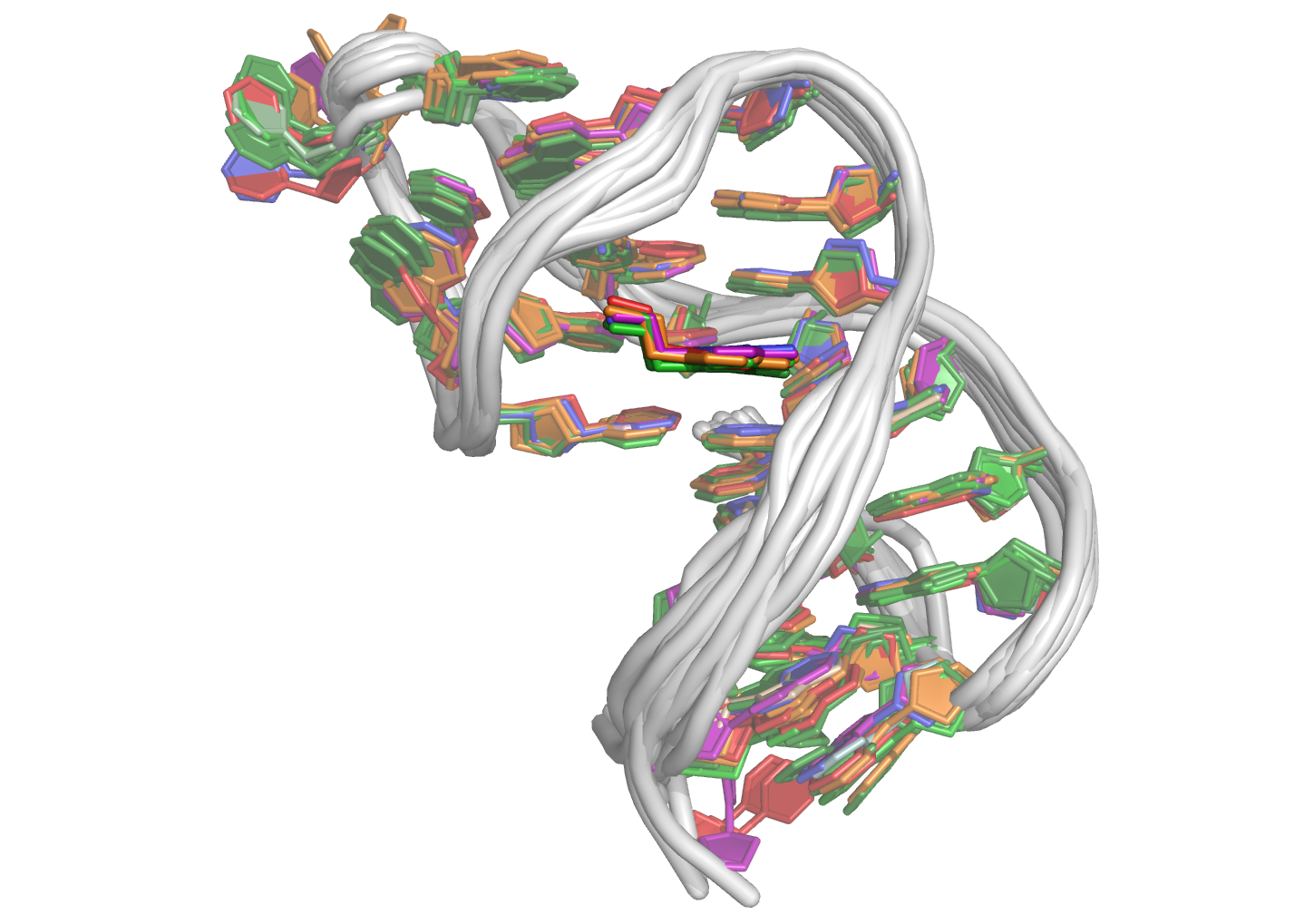
Figure 11: Fifteen minimized structures obtained by restrained molecular dynamics and minimization (left) and corresponding structures for the longer sequence calculated with and without A38•T37 base pairing. The dopamine ligand is shown in yellow, guanines in tan, thymines in green, adenines in blue and cytosine in purple.
The summary of the simulation energies is given in Table 1.
Table 2: Pairwise RMSD between the top 15 structures
2.2.2 PCA
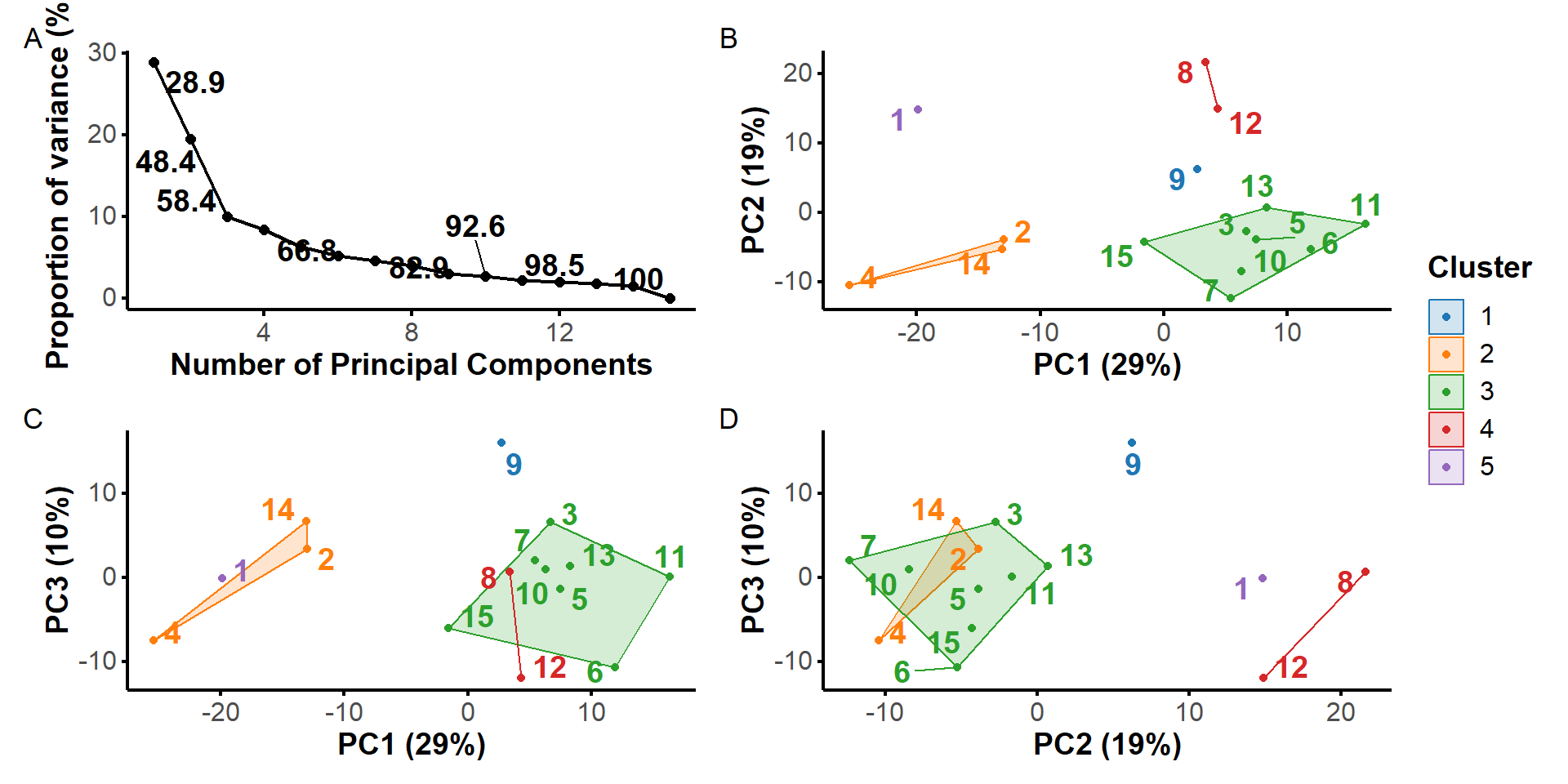
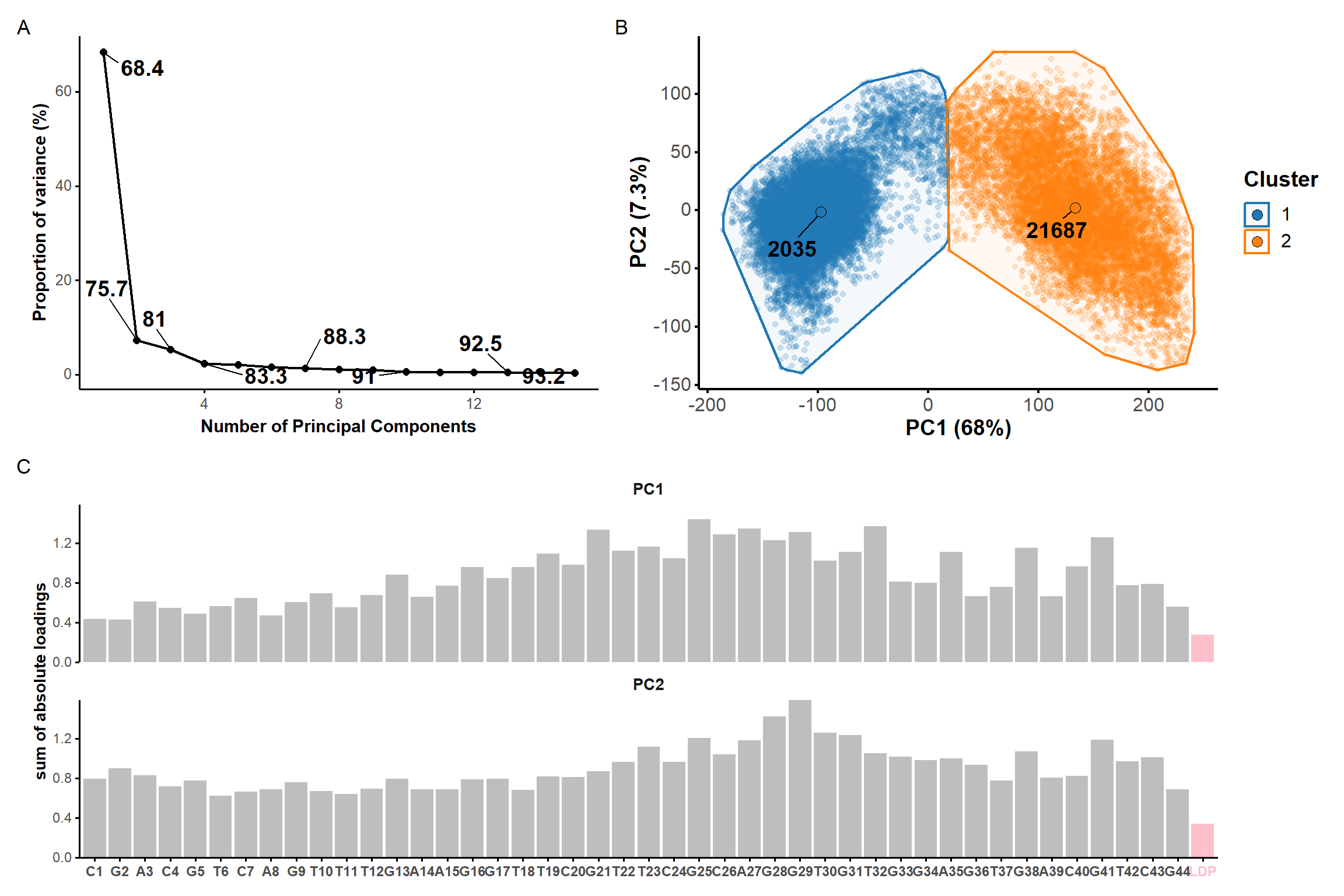
It is apparent that some structures deviate more from others, e.g. 1, 4, and 8 and, to a lower extent, 9,12 and 14. We aimed at identifying clusters of structures to summarize the conformational space explored by the aptamer. Below are shown the results of a principal component analysis based on all atoms except hydrogen atoms.
In [44]:
pca_pdb <-pca.pdbs( pdb, use.svd =FALSE, rm.gaps =TRUE, fit =FALSE#pdb models are already aligned at the import stage)scree <-data.frame(pc =1:length(pca_pdb$L),L = pca_pdb$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.), signif(cum.var, 3), NA) ) %>%ggplot(., aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' ) pca.plotr <-function(pca_object, dim.1, dim.2, max.pc =4, meth ="kmeans", dist ="euclidean", min_nc =3, max_nc =7,scale = scaling){# Run NbClust to get the best number of clusters and cluster assignment nb <-NbClust(data.frame(pca_object$z) %>%select(all_of(1:max.pc)), distance = dist, min.nc = min_nc, max.nc = max_nc, method = meth )# Extract and format scores scores <- pca_object$z %>%as_tibble() %>%set_names(paste0('PC', 1:ncol(.))) %>%mutate(State =1:n(), .before =1) %>%mutate(cluster = nb$Best.partition) %>%select(State, cluster, !!sym(dim.1), !!sym(dim.2))# print(scores)# Function to calculate convex hull coordinates for each cluster get_hull_coordinates <-function(cluster_data) { hull_indices <-chull(cluster_data$PC1, cluster_data$PC2) hull_coordinates <- cluster_data[hull_indices, ]return(hull_coordinates) }# Calculate hull coordinates for each cluster renamed_scores <- scores %>% magrittr::set_colnames(c('State', 'cluster', 'PC1', 'PC2')) hull_coordinates <-do.call(rbind, lapply(split(renamed_scores, renamed_scores$cluster), get_hull_coordinates)) %>% magrittr::set_colnames(c('State', 'cluster', dim.1, dim.2))# Calculate the variance explained by each PC var <-signif(pca_object$L/sum(pca_object$L) *100, 2)[c(as.numeric(gsub('PC', '', dim.1)), as.numeric(gsub('PC', '', dim.2)))]# Plot the scores and color by cluster assignment pca_plot <- hull_coordinates %>%ggplot(., aes_string(x = dim.1, y = dim.2)) +geom_polygon(aes(group = cluster, fill =factor(cluster), color =factor(cluster)),alpha =0.2 ) +geom_point(mapping =aes(color =factor(cluster)),data = scores ) +geom_text_repel(mapping =aes(label = State, color =factor(cluster)),data = scores,size =5,fontface ='bold',show.legend =FALSE ) +custom.theme(scale) +labs(x = glue::glue(dim.1, ' (', var[1], '%)'),y = glue::glue(dim.2, ' (', var[2], '%)') ) +scale_color_d3(name ='Cluster') +scale_fill_d3(name ='Cluster')return(pca_plot)}p.pca <- scree +pca.plotr(pca_pdb, 'PC1', 'PC2') +pca.plotr(pca_pdb, 'PC1', 'PC3') +pca.plotr(pca_pdb, 'PC2', 'PC3') +plot_layout(guides ='collect') &plot_annotation(tag_levels =c('A', 'B', 'C', 'D'))
Warning in pf(beale, pp, df2): NaNs produced
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 6 proposed 3 as the best number of clusters
* 3 proposed 4 as the best number of clusters
* 8 proposed 5 as the best number of clusters
* 4 proposed 6 as the best number of clusters
* 2 proposed 7 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 5
*******************************************************************
Warning: The `x` argument of `as_tibble.matrix()` must have unique column names if
`.name_repair` is omitted as of tibble 2.0.0.
ℹ Using compatibility `.name_repair`.
Warning: `aes_string()` was deprecated in ggplot2 3.0.0.
ℹ Please use tidy evaluation idioms with `aes()`.
ℹ See also `vignette("ggplot2-in-packages")` for more information.
Warning in pf(beale, pp, df2): NaNs produced
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 6 proposed 3 as the best number of clusters
* 3 proposed 4 as the best number of clusters
* 8 proposed 5 as the best number of clusters
* 4 proposed 6 as the best number of clusters
* 2 proposed 7 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 5
*******************************************************************
Warning in pf(beale, pp, df2): NaNs produced
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 6 proposed 3 as the best number of clusters
* 3 proposed 4 as the best number of clusters
* 8 proposed 5 as the best number of clusters
* 4 proposed 6 as the best number of clusters
* 2 proposed 7 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 5
*******************************************************************
Figure 12: Principal component analysis on the coordinates of the 15 minimized structures. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points, and (B) to (D): score plots along two dimensions for the first three principal components. Clusters determined by kmeans are colored.
`summarise()` has grouped output by 'resno', 'resid', 'PC'. You can override
using the `.groups` argument.
###
Figure 13: Sum of absolute loadings of residues for the first three first principal components
As expected from the analysis of trajectories, the key differences between the clusters are the loop thymine T23, the 3’end, and G25.
Minimized structures colored by clusters
2.2.3 NMR violations
In [47]:
rstslt.violations <-rst.error.batchr(input.file ='data/nmr_viol_doprstslt.txt', error.thresh =0.15, occurence.thresh =0, states =15)
── Column specification ────────────────────────────────────────────────────────
cols(
.default = col_character(),
X3 = col_double(),
X9 = col_double(),
X26 = col_double(),
X29 = col_double(),
X31 = col_double(),
X32 = col_double()
)
ℹ Use `spec()` for the full column specifications.
Warning: There were 15 warnings in `mutate()`.
The first warning was:
ℹ In argument: `state.1 = .Primitive("as.double")(state.1)`.
Caused by warning:
! NAs introduced by coercion
ℹ Run `dplyr::last_dplyr_warnings()` to see the 14 remaining warnings.
The average violations observed across the 20 minimized structures, with a cut-off of 0.15 Å are presented in Table 3. No violation exceeds a mean error of 0.25 Å. Out of the 10 violations above the cutoff, 8 are present in more than half of the structures.
The base pairs identified in the 20 minimized structures are named, where possible, in Table 4, and further described using three nomenclatures.
The Saenger nomeclature is a list of 28 (plus one added later) base pairs identified by Roman numerals (Saenger 1984; Gesteland, Cech, and Atkins 2006). A canonical Watson-Crick base pair is XIX for GC, and XX for AT.
The Leontis-Westhof classification (LEONTIS and WESTHOF 2001), latter expanded by Lemieux and Major (Lemieux 2002), describes base-pairing by considering whether H-bonds are established on the Watson-Crick, Hoogsteen or sugar edges, and what is the glycosidic orientation (cis/trans: c/t) of the nucleotides. A canonical Watson-Crick base pair would then be cWW. Note that the sugar edge has been defined with RNA in mind, and therefore with 2’-OH that are absent here. This sugar edge is absent for syn glycosidic dihedral angles (see section Section 2.3 below), which is only the case of G31 here.
The DSSR classification is similar to the Leontis-Westhof, as it indicates the cis/trans (c/t) orientation followed by the interacting edges, which can be Watson-Crick (W), major (M; ~Hoogsteen) and minor grooves (m; ~sugar). It additionally incorporate the +/- base ’face indication as in the Saenger nomenclature. Here, a canonical Watson-Crick base pair is called cW-W.
In [50]:
c(1:15) %>%# initialize structure numbersas.character() %>%# if the number is less than 10, add a leading zerosapply(function(x) {if (nchar(x) ==1) {paste0('0', x) } else { x } }) %>%# paste the urlsapply(function(x) {paste0('https://raw.githubusercontent.com/EricLarG4/EricLarG4.github.io/master/indiv_pdb/5_min_doprstslt', x, '.pdb') }) %>%# feed to dssr functionlapply(., dssr)
Table 4: Base pairs identified by DSSR in the 15 minimized structures. Abbreviations. +/-: the paired nucleotides have same/opposite faces (as in canonical antiparallel Watson-Crick pairing); WC and W: Watson-Crick pairing/edge; H: Hoogsteen edge; S: sugar edge; M/m: Major/minor groove edges; .: undefined edge; r: reverse; c/t: cis/trans orientation along the glycosidic bond.
Remarkably, the whole structure only contains two canonical Watson-Crick base pairs (C24-G29 and C26-G33; Figure 14 (a)). One GC (C20-G28) and two AT ((T12-A35 and T19-A27) base pairs have rWC configurations (Figure 14 (b)). Note that T19 is bound by the dopamine ligand as well (see Section 2.5).
(a) Watson-Crick GC base pairs
(b) Reverse Watson-Crick AT base pairs
Figure 14: Watson-Crick and reverse Watson-Crick base pairs
2.3 Dihedral angles
All residues are in the anti configuration except for G31 in syn (Table 5). G31 forms a trans Watson-Crick base pair with G21 (tW+W; Figure 15).
Dihedral angles are stable along the simulation (Figure 16). The same is observed during the unrestrained simulation, albeit with more variance (Figure 17).
In [56]:
scaling <-0.7dihedrals_rstslt <- chi.rstslt %>%group_by(residue) %>%mutate(chi =ifelse(chi >180, chi -360, chi),mean.chi =mean(chi),mod.chi =if_else( mean.chi <20& mean.chi >0& chi <0, chi +360,if_else( mean.chi <-160& chi >0, chi -360, chi ) ),mean.chi =mean(mod.chi),gba =case_when( mean.chi >60& mean.chi <90~'syn', mean.chi <-60| mean.chi >-180~'anti',TRUE~'other' ),residue =str_replace_all(residue, 'D|3_|5_|_', ''),#add html colors to the residue as function of their gbaresidue.label =case_when( gba =='syn'~paste0('<span style="color:tomato">', residue, '</span>'), gba =='anti'~paste0('<span style="color:grey10">', residue, '</span>'),TRUE~'<span style="color:steelblue4">other</span>' ),residue.number =as.numeric(str_extract(residue, '\\d+')) ) ggplot(dihedrals_rstslt, aes(chi, frame, color = gba)) +facet_wrap(~factor( residue.label,levels = dihedrals_rstslt %>%arrange(residue.number) %>%pull(residue.label) %>%unique() )) +geom_point(alpha =0.005) +scale_y_continuous(expand =c(0,0), breaks =c(99999)) +scale_x_continuous(limits =c(-180, 180), breaks =c(-90, 0, 90, 180)) +scale_color_manual(values =rev(c('tomato', 'grey10'))) + ggthemes::theme_pander() +theme(axis.text.x =element_text(size =12* scaling, face ='bold'),axis.text.y =element_blank(),axis.title.x =element_blank(),axis.title.y =element_blank(),axis.ticks =element_blank(),strip.text.x =element_markdown(size =16* scaling, face ='bold'),strip.text.y =element_markdown(size =16* scaling, face ='bold'),strip.background =element_rect(),panel.grid.major.x =element_line(linewidth =0.5* scaling, color ='grey', linetype ='dotted'),panel.grid.minor.x =element_blank(),panel.grid.major.y =element_line(linewidth =0.5* scaling, color ='grey', linetype ='dotted'),panel.grid.minor.y =element_blank(),panel.border =element_blank(),panel.background =element_blank(),panel.spacing.y =unit(1, 'lines'),panel.spacing.x =unit(2, 'lines'),plot.title =element_markdown(size =16* scaling, face ='bold', hjust =0.5),plot.subtitle =element_markdown(size =14* scaling),plot.caption =element_markdown(hjust =1, size =14* scaling),legend.position ='none' ) +coord_polar(clip ='off')##
Figure 16: Dihedral angles of all residues for the rMD, wrapped in [-180;180], along the trajectory (from center to outside of circle)
In [57]:
scaling <-0.7dihedrals_unrstslt <- chi.unrstslt %>%group_by(residue) %>%mutate(chi =ifelse(chi >180, chi -360, chi),mean.chi =mean(chi),mod.chi =if_else( mean.chi <20& mean.chi >0& chi <0, chi +360,if_else( mean.chi <-160& chi >0, chi -360, chi ) ),mean.chi =mean(mod.chi),gba =case_when( mean.chi >60& mean.chi <90~'syn', mean.chi <-60| mean.chi >-180~'anti',TRUE~'other' ),residue =str_replace_all(residue, 'D|3_|5_|_', ''),#add html colors to the residue as function of their gbaresidue.label =case_when( gba =='syn'~paste0('<span style="color:tomato">', residue, '</span>'), gba =='anti'~paste0('<span style="color:grey10">', residue, '</span>'),TRUE~'<span style="color:steelblue4">other</span>' ),residue.number =as.numeric(str_extract(residue, '\\d+')) ) ggplot(dihedrals_unrstslt, aes(chi, frame, color = gba)) +facet_wrap(~factor( residue.label,levels = dihedrals_unrstslt %>%arrange(residue.number) %>%pull(residue.label) %>%unique() )) +geom_point(alpha =0.005) +scale_y_continuous(expand =c(0,0), breaks =c(99999)) +scale_x_continuous(limits =c(-180, 180), breaks =c(-90, 0, 90, 180)) +scale_color_manual(values =rev(c('tomato', 'grey10'))) + ggthemes::theme_pander() +theme(axis.text.x =element_text(size =12* scaling, face ='bold'),axis.text.y =element_blank(),axis.title.x =element_blank(),axis.title.y =element_blank(),axis.ticks =element_blank(),strip.text.x =element_markdown(size =16* scaling, face ='bold'),strip.text.y =element_markdown(size =16* scaling, face ='bold'),strip.background =element_rect(),panel.grid.major.x =element_line(linewidth =0.5* scaling, color ='grey', linetype ='dotted'),panel.grid.minor.x =element_blank(),panel.grid.major.y =element_line(linewidth =0.5* scaling, color ='grey', linetype ='dotted'),panel.grid.minor.y =element_blank(),panel.border =element_blank(),panel.background =element_blank(),panel.spacing.y =unit(1, 'lines'),panel.spacing.x =unit(2, 'lines'),plot.title =element_markdown(size =16* scaling, face ='bold', hjust =0.5),plot.subtitle =element_markdown(size =14* scaling),plot.caption =element_markdown(hjust =1, size =14* scaling),legend.position ='none' ) +coord_polar(clip ='off')
Figure 17: Dihedral angles of all residues for the unrestrained MD, wrapped in [-180;180], along the trajectory (from center to outside of circle)
2.4 Puckers
Most nucleotides have their sugar adopting a C2’-endo (typical in B DNA) or close to it (C1’-exo, C3’-exo; Table 6, Figure 18). Among those, T23 also visits the C1’-endo pucker during a large portion of the simulation (Figure 20), and can be found in several of the minimized structures; however this is of little significance given that this residue does not form any intra- or inter-molecular contacts.
G25, whose amino group binds to the N7 of G28 (tm-M; Figure 21 (a)), is found in half the structures in C1’-exo or C2’-endo but adopts a variety of puckers in others. During the microsecond simulation, it mostly exist in C1’-exo, and only visits the other configurations for a short period of time.
T18, which is stacked on G17, is tightly bound by the dopamine ligand (Figure 21 (b)) and forms an off-plane tW-M base pair with G33, has a C1’-endo configuration in most conformers, which has been observed for dsDNA around intercalator drugs or when gaining increasing flexibility (A. H. J. Wang et al. 1987; Shen et al. 2011). It is therefore likely that the backbone of T18 adopts a non-canonical conformation to allow dopamine binding. It remains stable during the simulation.
G16, C20, and G34 mostly exist in the C4’-exo/O4’-endo configurations, which is not very common for DNA (Anosova et al. 2015). G16 pairs with the Hoogsteen face of G34 (Figure 21 (c); tW-M) and their puckering is quite unstable during the simulation (Figure 20). C20 is involved in a triplet made of a reverse Watson-Crick base pair with G28 (tW+W) and a single H-bond to T32 (tW-.; Figure 21 (d)).
G17 has an extremely varied pucker depending on the minimized structure (9 unique puckers observed!), and has a remarkably diffuse pucker angle and amplitude during the course of the microsecond simulation. G17 is involved in a non coplanar triplet with the Hoogsteen face of G33 (tW-M; stacked under the dopamine) and T11 (cW-m; Figure 21 (e)). It also binds to G34 (tW-M) and is stacked on G16, whose puckering is also fairly unstable.
A15 is the only nucleotide that mostly adopts a C4’-endo pucker. It forms an asymetric homopurine base pair with A35 (Figure 21 (f); tW-M) , and the sugar pucker is stable during the simulation (Figure 20).
Figure 19: Sugar puckering calculated with the Altona & Sundaralingam method, wrapped from 0 to 360 for the restrained simulation. The position of the points relative to the center/border of the circles scales with the pucker amplitude (the larger the closer to the border). The circled points represent the median pucker/amplitude. Calculation performed on 5000 evenly spaced frames.
Figure 20: Pucker angles of all residues for the rMD, along the trajectory (from center to outside of circle). Residues with relative standard deviation above 20% are labeled as flexible
(a) Sheared base pair between the sugar edge of G25 (left) and the Hoogsteen edge of G28 (tm-M)
(b) Binding of T18 to dopamine, G33 (tW-M), and stacking on G17
(c) Watson-Crick to Hoogsteen base pairing of G16 and G34 (tW-M)
(d) Reverse Watson-Crick base pair between C20 and G28 (tW+W) within a triplet with T32
(e) Triplet of G17 (foreground, top right), T11 (cW-m; background) and G33 (tW-M, foreground, top left) under the dopamine binding site, and above G34 (bottom left; tW-M with G16 and G17, tW+W with T11).
(f) tW-M base pairing of A15 (right) with A35 (left), and to a lower extent G36 (bottom)
Figure 21: Non-canonical base pairing leading to ‘weird’ puckers
In the unrestrained simulations, almost all nucleotides have their sugar adopting a C2’-endo (typical in B DNA) or close to it (C1’-exo; Figure 22). Only T18 adopts a C4’-exo configuration. The non-canonical puckers observed above are therefore imposed by the restraints. For the specific case of T18, the more RNA-like pucker observed in the unrestrained simulation still differs from the C1’-endo pucker of its restrained counterpart.
Figure 22: Sugar puckering calculated with the Altona & Sundaralingam method, wrapped from 0 to 360 for the unrestrained simulation. The position of the points relative to the center/border of the circles scales with the pucker amplitude (the larger the closer to the border). The circled points represent the median pucker/amplitude.
2.5 Dopamine binding
The dopamine is intercalated between T32 and G33/C16, and forms H-bond contacts with the O4s of T18 and T19 with its two hydroxy groups (O1, O2; Figure 25). The ammonium group (N1 forms two additional H-bonds, with the N3 of of A27 (and O4’; although not always canonically since the bond angle is sometimes below 120°; Figure 24) and the deoxyribose O4’ of G28.
The H-bonds between O1/O2 of the dopamine and T18/T19 are globally stable during the restrained simulation (Figure 23), in particular the short LDP:O2/T18:O4 (Figure 28). The ammonium group (N1) also establishes stable H-bonds with G28 and A27.
In absence of restraints, the H-bond with A27 and G28 are longer and less stable. Conversely, the bond to T19 is shorter and stable. H-bond angles are also significantly much less stable.
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
[1] "Processing pair: T11:N3-G33:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 33"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T11:N3-G34:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 34"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T12:N3-A35:N1"
[1] "Residues: T T A"
[1] "Numbers: 12 12 35"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N2-G36:N3"
[1] "Residues: G G G"
[1] "Numbers: 13 13 36"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N1-T37:O4'"
[1] "Residues: G G T"
[1] "Numbers: 13 13 37"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A15:N6-A35:N7"
[1] "Residues: A A A"
[1] "Numbers: 15 15 35"
[1] "Elements: N6 H61 N7" "Elements: N6 H62 N7"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N1-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-T11:O2"
[1] "Residues: G G T"
[1] "Numbers: 17 17 11"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N1-G33:N7"
[1] "Residues: G G G"
[1] "Numbers: 17 17 33"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 17 17 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T19:N3-A27:N1"
[1] "Residues: T T A"
[1] "Numbers: 19 19 27"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C20:N4-T32:O4"
[1] "Residues: C C T"
[1] "Numbers: 20 20 32"
[1] "Elements: N4 H41 O4" "Elements: N4 H42 O4"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G29:N3"
[1] "Residues: G G G"
[1] "Numbers: 21 21 29"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N1-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-T22:O4'"
[1] "Residues: C C T"
[1] "Numbers: 24 24 22"
[1] "Elements: N4 H41 O4'" "Elements: N4 H42 O4'"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-G29:O6"
[1] "Residues: C C G"
[1] "Numbers: 24 24 29"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G28:N7"
[1] "Residues: G G G"
[1] "Numbers: 25 25 28"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G29:O6"
[1] "Residues: G G G"
[1] "Numbers: 25 25 29"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C26:N4-G33:O6"
[1] "Residues: C C G"
[1] "Numbers: 26 26 33"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A27:N6-T19:O2"
[1] "Residues: A A T"
[1] "Numbers: 27 27 19"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-C20:N3"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N1-C20:O2"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N1 H1 O2"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-T32:O4"
[1] "Residues: G G T"
[1] "Numbers: 28 28 32"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-G21:N3"
[1] "Residues: G G G"
[1] "Numbers: 29 29 21"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N1-C24:N3"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-C24:O2"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N2-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-T30:O4'"
[1] "Residues: G G T"
[1] "Numbers: 31 31 30"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N1-C26:N3"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N2-C26:O2"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N1-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N1 H1 O4"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N2-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-T12:O2"
[1] "Residues: A A T"
[1] "Numbers: 35 35 12"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-A15:N1"
[1] "Residues: A A A"
[1] "Numbers: 35 35 15"
[1] "Elements: N6 H61 N1" "Elements: N6 H62 N1"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-G13:N3"
[1] "Residues: G G G"
[1] "Numbers: 36 36 13"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A14:O4'"
[1] "Residues: G G A"
[1] "Numbers: 36 36 14"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N1 H1 N1"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N2 H21 N1" "Elements: N2 H22 N1"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O1-T18:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 18"
[1] "Elements: O1 HO1 O4"
[1] "Processing hydrogen: HO1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O2-T18:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 18"
[1] "Elements: O2 HO2 O4"
[1] "Processing hydrogen: HO2"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O1-T19:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 19"
[1] "Elements: O1 HO1 O4"
[1] "Processing hydrogen: HO1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-A27:N3"
[1] "Residues: LDP LDP A"
[1] "Numbers: 38 38 27"
[1] "Elements: N1 HN11 N3" "Elements: N1 HN12 N3" "Elements: N1 HN13 N3"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-A27:O4'"
[1] "Residues: LDP LDP A"
[1] "Numbers: 38 38 27"
[1] "Elements: N1 HN11 O4'" "Elements: N1 HN12 O4'" "Elements: N1 HN13 O4'"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-G28:O4'"
[1] "Residues: LDP LDP G"
[1] "Numbers: 38 38 28"
[1] "Elements: N1 HN11 O4'" "Elements: N1 HN12 O4'" "Elements: N1 HN13 O4'"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
pair.dist.unrst <-lapply( pair.list.rst$pair, #recycling of same contacts than rst! atom.distr,pdb.input = pdb.unrst.long,span =0.01) %>%do.call(rbind, .)
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
pair.angle.unrst <-lapply( pair.list.rst$pair, #recycling of same contacts than rst! bond.angler,pdb.input = pdb.unrst.h.long, info = info.long, span =0.01, clean =TRUE, rectify =50) %>%do.call(rbind, .)
[1] "Processing pair: T11:N3-G33:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 33"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T11:N3-G34:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 34"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T12:N3-A35:N1"
[1] "Residues: T T A"
[1] "Numbers: 12 12 35"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N2-G36:N3"
[1] "Residues: G G G"
[1] "Numbers: 13 13 36"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N1-T37:O4'"
[1] "Residues: G G T"
[1] "Numbers: 13 13 37"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A15:N6-A35:N7"
[1] "Residues: A A A"
[1] "Numbers: 15 15 35"
[1] "Elements: N6 H61 N7" "Elements: N6 H62 N7"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N1-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-T11:O2"
[1] "Residues: G G T"
[1] "Numbers: 17 17 11"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N1-G33:N7"
[1] "Residues: G G G"
[1] "Numbers: 17 17 33"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 17 17 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T19:N3-A27:N1"
[1] "Residues: T T A"
[1] "Numbers: 19 19 27"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C20:N4-T32:O4"
[1] "Residues: C C T"
[1] "Numbers: 20 20 32"
[1] "Elements: N4 H41 O4" "Elements: N4 H42 O4"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G29:N3"
[1] "Residues: G G G"
[1] "Numbers: 21 21 29"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N1-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-T22:O4'"
[1] "Residues: C C T"
[1] "Numbers: 24 24 22"
[1] "Elements: N4 H41 O4'" "Elements: N4 H42 O4'"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-G29:O6"
[1] "Residues: C C G"
[1] "Numbers: 24 24 29"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G28:N7"
[1] "Residues: G G G"
[1] "Numbers: 25 25 28"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G29:O6"
[1] "Residues: G G G"
[1] "Numbers: 25 25 29"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C26:N4-G33:O6"
[1] "Residues: C C G"
[1] "Numbers: 26 26 33"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A27:N6-T19:O2"
[1] "Residues: A A T"
[1] "Numbers: 27 27 19"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-C20:N3"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N1-C20:O2"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N1 H1 O2"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-T32:O4"
[1] "Residues: G G T"
[1] "Numbers: 28 28 32"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-G21:N3"
[1] "Residues: G G G"
[1] "Numbers: 29 29 21"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N1-C24:N3"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-C24:O2"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N2-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-T30:O4'"
[1] "Residues: G G T"
[1] "Numbers: 31 31 30"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N1-C26:N3"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N2-C26:O2"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N1-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N1 H1 O4"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N2-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-T12:O2"
[1] "Residues: A A T"
[1] "Numbers: 35 35 12"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-A15:N1"
[1] "Residues: A A A"
[1] "Numbers: 35 35 15"
[1] "Elements: N6 H61 N1" "Elements: N6 H62 N1"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-G13:N3"
[1] "Residues: G G G"
[1] "Numbers: 36 36 13"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A14:O4'"
[1] "Residues: G G A"
[1] "Numbers: 36 36 14"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N1 H1 N1"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N2 H21 N1" "Elements: N2 H22 N1"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O1-T18:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 18"
[1] "Elements: O1 HO1 O4"
[1] "Processing hydrogen: HO1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O2-T18:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 18"
[1] "Elements: O2 HO2 O4"
[1] "Processing hydrogen: HO2"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:O1-T19:O4"
[1] "Residues: LDP LDP T"
[1] "Numbers: 38 38 19"
[1] "Elements: O1 HO1 O4"
[1] "Processing hydrogen: HO1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-A27:N3"
[1] "Residues: LDP LDP A"
[1] "Numbers: 38 38 27"
[1] "Elements: N1 HN11 N3" "Elements: N1 HN12 N3" "Elements: N1 HN13 N3"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-A27:O4'"
[1] "Residues: LDP LDP A"
[1] "Numbers: 38 38 27"
[1] "Elements: N1 HN11 O4'" "Elements: N1 HN12 O4'" "Elements: N1 HN13 O4'"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: LDP38:N1-G28:O4'"
[1] "Residues: LDP LDP G"
[1] "Numbers: 38 38 28"
[1] "Elements: N1 HN11 O4'" "Elements: N1 HN12 O4'" "Elements: N1 HN13 O4'"
[1] "Processing hydrogen: HN11"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN12"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: HN13"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 75000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
###
In [67]:
pair.dist.noldp <-lapply(#recycling of same contacts than rst!#but without LDP pair.list.rst$pair[!str_detect(pair.list.rst$pair, "LDP")], atom.distr,pdb.input = pdb.noldp.long,span =0.01) %>%do.call(rbind, .)
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
pair.angle.noldp <-lapply(#recycling of same contacts than rst!#but without LDP pair.list.rst$pair[!str_detect(pair.list.rst$pair, "LDP")], bond.angler,pdb.input = pdb.noldp.h.long, info = info.long, span =0.01, clean =TRUE, rectify =50) %>%do.call(rbind, .)
[1] "Processing pair: T11:N3-G33:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 33"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T11:N3-G34:O6"
[1] "Residues: T T G"
[1] "Numbers: 11 11 34"
[1] "Elements: N3 H3 O6"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T12:N3-A35:N1"
[1] "Residues: T T A"
[1] "Numbers: 12 12 35"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N2-G36:N3"
[1] "Residues: G G G"
[1] "Numbers: 13 13 36"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G13:N1-T37:O4'"
[1] "Residues: G G T"
[1] "Numbers: 13 13 37"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A15:N6-A35:N7"
[1] "Residues: A A A"
[1] "Numbers: 15 15 35"
[1] "Elements: N6 H61 N7" "Elements: N6 H62 N7"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N1-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:N7"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G16:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 16 16 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-T11:O2"
[1] "Residues: G G T"
[1] "Numbers: 17 17 11"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N1-G33:N7"
[1] "Residues: G G G"
[1] "Numbers: 17 17 33"
[1] "Elements: N1 H1 N7"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G17:N2-G34:O6"
[1] "Residues: G G G"
[1] "Numbers: 17 17 34"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: T19:N3-A27:N1"
[1] "Residues: T T A"
[1] "Numbers: 19 19 27"
[1] "Elements: N3 H3 N1"
[1] "Processing hydrogen: H3"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C20:N4-T32:O4"
[1] "Residues: C C T"
[1] "Numbers: 20 20 32"
[1] "Elements: N4 H41 O4" "Elements: N4 H42 O4"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G29:N3"
[1] "Residues: G G G"
[1] "Numbers: 21 21 29"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N1-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G21:N2-G31:O6"
[1] "Residues: G G G"
[1] "Numbers: 21 21 31"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-T22:O4'"
[1] "Residues: C C T"
[1] "Numbers: 24 24 22"
[1] "Elements: N4 H41 O4'" "Elements: N4 H42 O4'"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C24:N4-G29:O6"
[1] "Residues: C C G"
[1] "Numbers: 24 24 29"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G28:N7"
[1] "Residues: G G G"
[1] "Numbers: 25 25 28"
[1] "Elements: N2 H21 N7" "Elements: N2 H22 N7"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G25:N2-G29:O6"
[1] "Residues: G G G"
[1] "Numbers: 25 25 29"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: C26:N4-G33:O6"
[1] "Residues: C C G"
[1] "Numbers: 26 26 33"
[1] "Elements: N4 H41 O6" "Elements: N4 H42 O6"
[1] "Processing hydrogen: H41"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H42"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A27:N6-T19:O2"
[1] "Residues: A A T"
[1] "Numbers: 27 27 19"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-C20:N3"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N1-C20:O2"
[1] "Residues: G G C"
[1] "Numbers: 28 28 20"
[1] "Elements: N1 H1 O2"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G28:N2-T32:O4"
[1] "Residues: G G T"
[1] "Numbers: 28 28 32"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-G21:N3"
[1] "Residues: G G G"
[1] "Numbers: 29 29 21"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N1-C24:N3"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G29:N2-C24:O2"
[1] "Residues: G G C"
[1] "Numbers: 29 29 24"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N1 H1 O6"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N2-G21:O6"
[1] "Residues: G G G"
[1] "Numbers: 31 31 21"
[1] "Elements: N2 H21 O6" "Elements: N2 H22 O6"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G31:N1-T30:O4'"
[1] "Residues: G G T"
[1] "Numbers: 31 31 30"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N1-C26:N3"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N1 H1 N3"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G33:N2-C26:O2"
[1] "Residues: G G C"
[1] "Numbers: 33 33 26"
[1] "Elements: N2 H21 O2" "Elements: N2 H22 O2"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N1-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N1 H1 O4"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G34:N2-T11:O4"
[1] "Residues: G G T"
[1] "Numbers: 34 34 11"
[1] "Elements: N2 H21 O4" "Elements: N2 H22 O4"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-T12:O2"
[1] "Residues: A A T"
[1] "Numbers: 35 35 12"
[1] "Elements: N6 H61 O2" "Elements: N6 H62 O2"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: A35:N6-A15:N1"
[1] "Residues: A A A"
[1] "Numbers: 35 35 15"
[1] "Elements: N6 H61 N1" "Elements: N6 H62 N1"
[1] "Processing hydrogen: H61"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H62"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-G13:N3"
[1] "Residues: G G G"
[1] "Numbers: 36 36 13"
[1] "Elements: N2 H21 N3" "Elements: N2 H22 N3"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A14:O4'"
[1] "Residues: G G A"
[1] "Numbers: 36 36 14"
[1] "Elements: N1 H1 O4'"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N1-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N1 H1 N1"
[1] "Processing hydrogen: H1"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 25000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
[1] "Processing pair: G36:N2-A15:N1"
[1] "Residues: G G A"
[1] "Numbers: 36 36 15"
[1] "Elements: N2 H21 N1" "Elements: N2 H22 N1"
[1] "Processing hydrogen: H21"
Union of selects
* Selected a total of: 3 atoms *
[1] "Processing hydrogen: H22"
Union of selects
* Selected a total of: 3 atoms *
[1] "Length of angle list before cleaning: 50000"
[1] "Length of angle list after cleaning: 25000"
[1] "Finished angle list nrow 25000"
Figure 23: Lengths of H-bonds (donor-acceptor distance) involved in dopamine binding during the restrained (top) and unrestrained (bottom) simulations. The lines are the result of local polynomial regression fitting with a span of 0.01. The canonical C26•G33 bp was used as a reference.
Figure 24: Angles of H-bonds (donor-H-acceptor atoms) established by the dopamine ligand during the microsecond simulation, with the canonical C26•G33 bp used as a reference. When multiple interconverting hydrogen atoms are involved, all are considered and each data point reflect the angle formed by the hydrogen most likely to establish an H-bond. The lines are the result of local polynomial regression fitting with a span of 0.01. The angle stays below 140 ° during the simulation in the case of A27:O4’, which is suboptimal to establish an H-bond. Note that the hydrogen atoms interconvert frequently during the simulation; all three atoms were considered and .
T18 is off-plane as it also binds to the Hoogsteen face of G33 stacked ‘under’ the dopamine. G33 is itself base pairing canonically to C26 (see also Figure 14 (a)).
Conversely, T19 is coplanar to the dopamine, and the two form a triplet with A27. T19 and A27 are arranged in a reverse WC configuration (see also Figure 14 (b)).
(a) Binding site of dopamine
(b) 2D interaction diagram. The ligand is shown in yellow, residues in orange, water in light blue, carbon in black, nitrogen in blue and oxygen in red. Hydrogen bonds are shown as dashed green lines. Hydrophobic interactions are highlighted with red lines.
Figure 25: Dopamine binding in the minimized structures obtained by rMD.
In absence of NMR restraints, we noticed that the ammonium side-chain flips between configurations ‘above’ or ‘below’ the catechol plane. We performed PCA on the binding pocket (T18:T19-C26:G28-T32:G33 and dopamine) to identify frames representative of the different configurations.
In [70]:
# Create a binding pocket trimmed pdb object# from the long unrestrained simulationcore.pdb <- pdb.unrst.long %>%trim.pdb( .,atom.select( .,resno =c(18, 19, 26, 27, 28, 32, 33, 38) ) )# Re-align frames to the binding pocket onlycore.pdb$xyz <-fit.xyz(fixed = core.pdb$xyz[1,],mobile = core.pdb$xyz,fixed.inds =atom.select(core.pdb, elety =unique(core.pdb$atom$elety))$xyz,mobile.inds =atom.select(core.pdb, elety =unique(core.pdb$atom$elety))$xyz)# Perform PCApca_unrst_core <-pca.pdbs( core.pdb, use.svd =FALSE, rm.gaps =TRUE, fit =FALSE#pdb models are already aligned at the import stage)# Scree plotscree_unrst_core <-data.frame(pc =1:length(pca_unrst_core$L),L = pca_unrst_core$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.), signif(cum.var, 3), NA) ) %>%ggplot(., aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' ) # NbClust determination [LONG!]pca.nb.unrst <-NbClust(data.frame(pca_unrst_core$z) %>%select(all_of(1:4)),distance ="euclidean",min.nc =3, max.nc =5,method ="kmeans")
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 5 proposed 3 as the best number of clusters
* 15 proposed 4 as the best number of clusters
* 3 proposed 5 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 4
*******************************************************************
# count the number of times each number of clusters was selected as the besttbl.clusters <-table(pca.nb.unrst$Best.partition)frac.clust.1<-round(tbl.clusters[1]/sum(tbl.clusters)*100, 1)frac.clust.4<-round(tbl.clusters[4]/sum(tbl.clusters)*100, 1)# plot loadingsp.loads.unrst <-data.frame(u =c(pca_unrst_core$U[,1], pca_unrst_core$U[,2], pca_unrst_core$U[,3]),eleno =rep(core.pdb$atom$eleno, 3),PC =c(rep('PC1', length(pca_unrst_core$U[,1])),rep('PC2', length(pca_unrst_core$U[,2])),rep('PC3', length(pca_unrst_core$U[,3])))) %>%left_join( core.pdb$atom,by =c("eleno"="eleno") ) %>%group_by(resno, resid, PC, chain) %>%summarise(u =sum(abs(u))) %>%mutate(resno =if_else(resid =='LDP', 38, resno),resid =gsub('^D', '', resid),label =if_else( resid =='LDP', #make LDP bold and pinkpaste0('<span style="color:pink"><b>', resid, '</b></span>'),paste0('<b>', resid, resno, '</b>') ) ) %>%filter(PC %in%paste0('PC', 1:2)) %>%ggplot(aes(x =factor( resno,levels =unique(resno),labels =unique(label) ),y = u,fill = chain ) ) +facet_wrap(~PC, ncol =1) +geom_bar(stat ="identity") +scale_fill_manual(values =c('grey', 'pink')) +scale_y_continuous(expand =c(0, 0)) +custom.theme(1) +theme(axis.title.x =element_blank(),legend.position ='none') +labs(y ='sum of absolute loadings')
`summarise()` has grouped output by 'resno', 'resid', 'PC'. You can override
using the `.groups` argument.
#full plotp.pca.unrst <- scree_unrst_core +pca.plotr.traj(pca_unrst_core, 'PC1', 'PC2', nb = pca.nb.unrst) + p.loads.unrst +plot_layout(design =' AB CC ') &plot_annotation(tag_levels =c('A', 'B', 'C', 'D'))
Figure 26: Principal component analysis on the coordinates of the binding pocket in the unrestrained microsecond trajectory. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points. (B) Score plots along the first two principal components, colored by kmeans clusters. (C) Sum of absolute loadings of residues for the first two first principal components
Four clusters were identified and the structures closest to their centroids are shown in Figure 27. The main difference if the configuration of the ammonium chain. The ammonium binds to either:
The O4’ and N3 of G33, which is stacked under the dopamine (top-left structure)
the O2 of T32, which is stacked above the dopamine (top-right structure)
the O4’ and N3 of G28 (bottom-left structure)
the O2 of T32 and N3 of G28, in a configuration midway between the two above (bottom-right structure)
The first configuration only accounts for 2.6% of the simulation. In the three latter case, T32 overlaps more with the catechol moiety, which increases \(\pi-\pi\) interactions. The fourth configuration is the more frequent (41.9% of the frames are in this cluster).
Neither of the four configurations follows exactly the restrained one. The ‘above’ configurations are closer to it, but the ammonium binds differently in each three case. It is folded back above the chain (akin to a scorpio) and does not bind to A27. However, beyond these four discrete examples, we evidence below that the ammonium is still able to bind to A27:N3 in around of third of the frames (Figure 28). For reference, it is almost always bound when restrained (Figure 28).
Figure 27: Binding modes in absence of NMR restraints. Four main binding modes have been evidenced by PCA, mostly varying by the configuration of the amino chain.
2.6 H-bond formation and stability
2.6.1 Complex
Besides those from the binding site, the formation of all H-bonds was monitored over the course of the simulation. Below, the list of monitored bonds is based on H-bonds detected in the minimized structures obtained by rMD; therefore Figure 28 ignores by design bonds formed exclusively in the uMD simulation and/or in absence of dopamine. The purpose of Figure 28 is therefore to establish the stability of H-bonds during the rMD simulation and assess whether they ‘survived’ in absence of restraints and dopamine.
The tight binding of dopamine to T18:O4 by the catechol moiety is confirmed. It is still present in absence of restraints but less frequently. The ammonium group almost always bind the sugar of G28 and the N3 of A27. Similarly, the frequency of H-bond formation is decreased in absence of restraints.
The less canonical binding to the sugar of the A27 is also observed, with low frequency but across the whole simulation, in the rMD. It is almost absent in absence of restraints, as seen above.
Conversely to the observations above, the H-bonding of dopamine to T19 is less frequent and tight in the rMD than in the uMD, as expected from bond length analysis above (Figure 23).
Figure 28: Formation of H-bonds over the course of the simulations with restraints (rMD), without restraints (uMD) and unrestrained with no dopamine (no LDP). The list of H-bonds was established on the minimized structures from the rMD simulations, with a maximum donor-acceptor distance of maximum 3.1 Å, keeping those formed in at least 20% of structures. H-bonds in the 5’- and 3’-end regions (residues 11:17 and 33:37() are colored in blue, and those involving the dopamine in pink. Top: each line is a frame (one frame every 0.04 ns); the darker a frame, the shorter and more linear the H-bond (the transparency follows linearly the bond angle/length ratio). Bottom: Cumulative frequencies. The frequency of bond formation is shown for relaxed (3.5 Å and 120°; light colors) and more stringent (3.0 Å and 140°; dark colors) cut-offs. Only inter-residue H-bonds with a ‘stringent’ frequency above 0.10 are shown.
2.6.2 (Pre-folded) free DNA
Generally speaking, the residues in the 5’ and 3’-end regions (in blue in Figure 28) are remarkably well conserved in absence of dopamine, when starting the simulation from the folded structure. This may indicate that the oligonucleotide can prefold into a short stem intermediate before the binding occurs between this stem and the very dynamic loops, after which the latter could finish folding.
PCA on the trajectory yielded three clusters (Figure 29), whose centroid coordinates are shown in Figure 30. The terminus region can be nicely aligned across clusters, but also against the complex, further demonstrating that the stem is very stable during the simulation, and retains the complex structure.
The loop region also maintains the same local folding in all clusters (it can be locally aligned), however its orientation relative to the terminus stem changes dramatically. It can be orthogonal (cluster 1; Figure 30; left), yielding a linear configuration (‘open’), or parallel, forming a more globular structure (‘closed’), closer to that of the complex (cluster 3; Figure 30; right). The second cluster contain intermediate conformations between these two extrema (cluster 2; Figure 30; center).
In [74]:
#PCApca_noldp <-pca.pdbs( pdb.noldp.long, use.svd =FALSE, rm.gaps =TRUE, fit =FALSE#pdb models are already aligned at the import stage)scree <-data.frame(pc =1:length(pca_noldp$L),L = pca_noldp$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.), signif(cum.var, 3), NA) ) %>%ggplot(., aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' )
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 11 proposed 3 as the best number of clusters
* 8 proposed 4 as the best number of clusters
* 5 proposed 5 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 3
*******************************************************************
Figure 29: Principal component analysis on the coordinates of the unbound oligonucleotide in the unrestrained microsecond trajectory. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points. (B) Score plots along the first two principal components, colored by kmeans clusters. (C) Sum of absolute loadings of residues for the first two first principal components
In [78]:
#create pdb objectpdb.noldp.pca <- pdb.noldp.h.long#keep the coordinates of the centroids of the clusterspdb.noldp.pca$xyz <- pdb.noldp.pca$xyz[c(4453, 14840, 20352),]#align on the stempdb.noldp.pca$xyz <-fit.xyz(fixed = pdb.noldp.pca$xyz[1,],mobile = pdb.noldp.pca$xyz,fixed.inds =atom.select( pdb.noldp.pca, resno =c(11:16, 34:36) )$xyz,mobile.inds =atom.select( pdb.noldp.pca,resno =c(11:16, 34:36) )$xyz)#write pdb fileif (!file.exists('data/pdb.noldp.pca.pdb')) {write.pdb(pdb.noldp.pca, file ='data/pdb.noldp.pca.pdb')}
Figure 30: Structures of the centroids of each cluster (1 to 3 from left to right) superimposed with the first minimized structure of the complex (grey).
In [79]:
tbl.clusters.noldp <-table(pca.nb.noldp$Best.partition)frac.clust.no.ldp.1<-round(tbl.clusters.noldp[1]/sum(tbl.clusters.noldp)*100, 1)frac.clust.no.ldp.2<-round(tbl.clusters.noldp[2]/sum(tbl.clusters.noldp)*100, 1)frac.clust.no.ldp.3<-round(tbl.clusters.noldp[3]/sum(tbl.clusters.noldp)*100, 1)open.times <-tibble(frame =1:length(pca.nb.noldp$Best.partition),cluster = pca.nb.noldp$Best.partition) %>%#create a group column that increments every time the cluster changesmutate(group =cumsum(c(1, diff(cluster) !=0)),t = frame * info.long$time.per.frame ) %>%group_by(cluster, group) %>%#calculate the time spent in each clustersummarise(t =max(t) -min(t) ) %>%group_by(cluster) %>%summarise(max.t =signif(max(t), 2),mean.t =signif(mean(t), 2) )
`summarise()` has grouped output by 'cluster'. You can override using the
`.groups` argument.
The unbound aptamer spends most of its time in the ‘closed’ (43.8%) and ‘intermediate’ (43.5%) configurations, and only occasionally visits the ‘open’, linear conformation (12.7%) (Figure 31). The ‘closed’ conformation has a mean lifetime of 0.7 ns and a max residency time of 51 ns. The ‘open’ configuration has a lower mean lifetime 0.54 ns, and is never visited more than 24 ns. The so-called ‘intermediate’ conformation is aptly named as it has an even lower mean lifetime 0.5 ns, and visits never exceed 9.2 ns.
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element C1' to C
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element C1' to C
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element C1' to C
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element C1' to C
Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: span too small. fewer data values than degrees of freedom.
Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: pseudoinverse used at 0.99
Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: reciprocal condition number 0
Warning in simpleLoess(y, x, w, span, degree = degree, parametric = parametric,
: There are other near singularities as well. 4.0401
###
In [81]:
p.open
Figure 31: Opening/closing of the unbound oligonucleotide during the microsecond simulation. Top: Cluster assignment for each frame. Bottom: Distance between the C1’ atoms of C24 (loop region) and A35 (stem region). The distance measured for each cluster centroid is shown as a dashed line.
2.7 Long aptamer
The long aptamer was studied from starting structure including (doplong8) or devoid of (doplong7) base pairing between A8 and T37. The minimized structures shown in Figure 11 were obtained by minimizing the last frame of 10-ns *restrained* simulations. The dynamics of these systems was explored with 1 microsecond unrestrained simulations.
The RMSD (vs. the first frame) is quickly plateauing when the A38•T37 is pre-formed (and restrained), but never does in absence of this base pair (Figure 32, Figure 33).
In [82]:
short.longdop.traj <-list('doplong7'= doplong7.traj, 'doplong8'= doplong8.traj)lapply(seq_along(short.longdop.traj),function(x) {data.frame(rmsd =rmsd(a = short.longdop.traj[[x]]$xyz[1,],b = short.longdop.traj[[x]]$xyz[seq(1, nrow(short.longdop.traj[[x]]$xyz), by =1), ],fit =FALSE ),frame = (seq(1, nrow(short.longdop.traj[[x]]$xyz), by =1)) #divider set to 1 to keep all frames ) %>%mutate(t = frame * info.short$time.per.frame,traj = x) }) %>%do.call(rbind, .) %>%mutate(traj =case_when( traj ==1~'doplong7 (no 8:37)', traj ==2~'doplong8 (with 8:37)' )) %>%ggplot(aes(x = t, y = rmsd, color = traj)) +geom_line() +custom.theme(1) +labs(x ='t (ns)',y ='RMSD (Å)', ) +scale_color_manual(values =c('#e62e73', '#003e83')) ###
Figure 32: RMSD on all residues (including 5’ and 3’-ends) without hydrogens across the 10 ns restrained simulation of the long aptamer, where the first frame is used as reference.
In [83]:
rmsd.pairwise.doplong7 <-rmsd(a = doplong7.traj$xyz[seq(1, nrow(doplong7.traj$xyz), by =nrow(doplong7.traj$xyz)/1000),], #keep one out of 5 framesfit =FALSE) %>%lazy_dt() %>%mutate(var1 =1:nrow(.), .before =1) %>%pivot_longer(cols =-var1, names_to ="var2", values_to ="value") %>%mutate(var2 =as.numeric(gsub("V", "", var2))) %>%mutate(traj ='doplong7') %>%as.data.table()
Warning in rmsd(a = doplong7.traj$xyz[seq(1, nrow(doplong7.traj$xyz), by = nrow(doplong7.traj$xyz)/1000), : No indices provided, using the 1431 non NA positions
rmsd.pairwise.doplong8 <-rmsd(a = doplong8.traj$xyz[seq(1, nrow(doplong8.traj$xyz), by =nrow(doplong8.traj$xyz)/1000),], #keep one out of 5 framesfit =FALSE) %>%lazy_dt() %>%mutate(var1 =1:nrow(.), .before =1) %>%pivot_longer(cols =-var1, names_to ="var2", values_to ="value") %>%mutate(var2 =as.numeric(gsub("V", "", var2))) %>%mutate(traj ='doplong8') %>%as.data.table()
Warning in rmsd(a = doplong8.traj$xyz[seq(1, nrow(doplong8.traj$xyz), by = nrow(doplong8.traj$xyz)/1000), : No indices provided, using the 1431 non NA positions
Figure 33: All-to-all RMSD (all residues and ligand atoms except H) for the 10 ns restrained simulation of the long aptamer.
2.7.1.2 Unrestrained microsecond simulations
In absence of restraints, doplong7 settles at RMSD values close to that of the final frame from the restrained 10 ns simulation (Figure 34). The pairwise data also reveals that an event clearly occurs after 56 ns (Figure 35), that is close to the start of the microsecond simulation but past the endpoint of the restrained 10 ns simulation. After this conformational change, the pairwise RMSD remains apparently stable, but other changes still occur (see below).
In presence of the pre-formed A38•T37 (doplong8), the simulation plateaus much faster (Figure 34)., as seen above for the retrained production run, but at about twice the RMSD. A very significant event is visible at 576 ns, resulting in a doubled RMSD vs. the first frame. The RMSD then varies much more significantly than before, with another significant change at 663 ns.
In [85]:
doplong7_longtraj.traj <-list('doplong7'= doplong7_longtraj, 'doplong8'= doplong8_longtraj)lapply(seq_along(doplong7_longtraj.traj),function(x) {data.frame(rmsd =rmsd(a = doplong7_longtraj.traj[[x]]$xyz[1,],b = doplong7_longtraj.traj[[x]]$xyz[seq(1, nrow(doplong7_longtraj.traj[[x]]$xyz), by =1), ],fit =FALSE ),frame = (seq(1, nrow(doplong7_longtraj.traj[[x]]$xyz), by =1)) #divider set to 1 to keep all frames ) %>%mutate(t = frame * info.long$time.per.frame,traj = x) }) %>%do.call(rbind, .) %>%mutate(traj =case_when( traj ==1~'doplong7 (no 8:37)', traj ==2~'doplong8 (with 8:37)' )) %>%ggplot(aes(x = t, y = rmsd, color = traj)) +geom_line() +custom.theme(1) +labs(x ='t (ns)',y ='RMSD (Å)', ) +scale_color_manual(values =c('doplong7 (no 8:37)'='#e62e73', 'doplong8 (with 8:37)'='#003e83')) +geom_segment(inherit.aes =FALSE,data =data.frame(traj =c(rep('doplong7 (no 8:37)', 3), rep('doplong8 (with 8:37)', 2)),x =c(56.5, 375, 553, 576, 663),y =c(13, 13.5, 14, 14, 15) ),aes(x = x, xend = x,y = y, yend = (y -1),color = traj ),arrow =arrow(type ="closed", length =unit(0.02, "npc")),linewidth =1 )
Figure 34: RMSD on all residues (including 5’ and 3’-ends) without hydrogens across the microsecond unrestrained simulation of the long aptamer, where the first frame is used as reference. The arrows point to conformational changes that are not all evident from this metric alone.
In [86]:
rmsd.pairwise.doplong7_longtraj <-rmsd(a = doplong7_longtraj$xyz[seq(1, nrow(doplong7_longtraj$xyz), by =nrow(doplong7_longtraj$xyz)/1000),], #keep one out of 5 framesfit =FALSE) %>%lazy_dt() %>%mutate(var1 =1:nrow(.), .before =1) %>%pivot_longer(cols =-var1, names_to ="var2", values_to ="value") %>%mutate(var2 =as.numeric(gsub("V", "", var2))) %>%mutate(traj ='doplong7') %>%as.data.table()
Warning in rmsd(a = doplong7_longtraj$xyz[seq(1, nrow(doplong7_longtraj$xyz), : No indices provided, using the 921 non NA positions
rmsd.pairwise.doplong8_longtraj <-rmsd(a = doplong8_longtraj$xyz[seq(1, nrow(doplong8_longtraj$xyz), by =nrow(doplong8_longtraj$xyz)/1000),], #keep one out of 5 framesfit =FALSE) %>%lazy_dt() %>%mutate(var1 =1:nrow(.), .before =1) %>%pivot_longer(cols =-var1, names_to ="var2", values_to ="value") %>%mutate(var2 =as.numeric(gsub("V", "", var2))) %>%mutate(traj ='doplong8') %>%as.data.table()
Warning in rmsd(a = doplong8_longtraj$xyz[seq(1, nrow(doplong8_longtraj$xyz), : No indices provided, using the 921 non NA positions
Figure 35: All-to-all RMSD (all residues and ligand atoms except H) for the unrestrained microsecond simulation. The white arrows point to significant events. The arrows point to conformational changes that are not all evident from this metric alone.
2.7.2 Base pairing
2.7.2.1 A38•T37 formation
Analysis of base pairing along the unrestrained simulation provides a straightforward explanation for the large conformational change of doplong7: the A8 and T37, initially far apart, rapidly base pair (canonical cW-W). This is accompanied by the formation of a base pair between G9 and the Hoogsteen face of G13. The latter is also rapidly formed in doplong8, wherein the A38•T37 bp is initially formed; however the base pairing is ‘inverted’:
doplong7: tW-M (VII in Saenger nomenclature), with G9•N2-G13:O6 and G9•N1-G13-N7 H-bonds
doplong8: a cW+M (VI in Saenger nomenclature), with G9•N2-G13-N7 and G9•N1-G13:O6 H-bonds
Note that this base pair is fairly unstable in doplong8, but A38•T37 remains formed nonetheless (reminder: there is no restraint here).
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N7 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N6 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N4 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O6 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N2 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O1 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4 to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N3 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O4' to O
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element N1 to N
Warning in atom2ele.default(x, elety.custom, rescue, ...):
mapped element O2 to O
Figure 36: Length of potential A38•T37, and G9•G13 H-bonds during the simulation. Note that the G9•G13 pairing differs. The pink arrow shows the onset of basepairing for doplong7.
2.7.2.2 Other events
The above does not explain what happens in the second part of doplong8’s simulation, nor is the only event occuring for doplong7.
doplong7: Two discrete events occur at around 375 and 553 ns
First, two of the three H-bonds from C24•G29 bonds get disrupted
Then the third gives up as well. The neighboring G25•G28 base pair is also disrupted at this last onset (Figure 40).
doplong8: Two discrete events as well, at around 576 and 663 ns.
First occurs the formation of the consecutive A15•A35, G16•G34 and G17•G33 base pairs in lieu of G34•T11, which is the expected base pair from NMR data of the short aptamer.
Second, the C24•G29 and G25•G28 base pairs are lost, similar to doplong7 (Figure 40).
Note also the transient flipping out of G9 after these events. The G36 base pair remains in contact, although it now adopts a co-planar configuration.
All of these conformational changes occur within the region of the aptamer present in the smaller version, and are very likely to be the consequence of the absence of retraints in this microsecond simulation; since all lost base pairs are present in the restrained simulations.
Figure 37: H-bond lengths for selected nucleotides undergoing base pair formation/disruption during the unrestrained microsecond simulation. The vertical lines indicate the onset of formation/disruption and correspond to the arrows in the figures above.
2.7.3 PCA
Principal component analysis allows to extract coordinates characteristic of the conformers prior and after the conformational changes.
In [91]:
pca_doplong7_longtraj <-pca.pdbs( doplong7_longtraj,use.svd =FALSE,rm.gaps =TRUE,fit =FALSE#pdb models are already aligned at the import stage)
In [92]:
pca_doplong8_longtraj <-pca.pdbs( doplong8_longtraj,use.svd =FALSE,rm.gaps =TRUE,fit =FALSE#pdb models are already aligned at the import stage)
In [93]:
scree7 <-data.frame(pc =1:length(pca_doplong7_longtraj$L),L = pca_doplong7_longtraj$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.),signif(cum.var, 3), NA) ) %>%ggplot(., aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' )scree8 <-data.frame(pc =1:length(pca_doplong8_longtraj$L),L = pca_doplong8_longtraj$L) %>%mutate(var = L/sum(L) *100,cum.var =cumsum(var) ) %>%filter(pc <=15) %>%select(-L) %>%mutate(label =ifelse( pc %in%1:3| pc %in%seq(4, nrow(.), 3) | pc ==nrow(.),signif(cum.var, 3), NA) ) %>%ggplot(aes(x = pc, y = var)) +geom_text_repel(aes(label = label), size =5, fontface ='bold', force =100) +geom_line(linewidth =0.75) +geom_point(size =2) +custom.theme(scaling) +labs(x ='Number of Principal Components',y ='Proportion of variance (%)' )
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 10 proposed 2 as the best number of clusters
* 7 proposed 3 as the best number of clusters
* 4 proposed 4 as the best number of clusters
* 3 proposed 5 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 2
*******************************************************************
*** : The Hubert index is a graphical method of determining the number of clusters.
In the plot of Hubert index, we seek a significant knee that corresponds to a
significant increase of the value of the measure i.e the significant peak in Hubert
index second differences plot.
*** : The D index is a graphical method of determining the number of clusters.
In the plot of D index, we seek a significant knee (the significant peak in Dindex
second differences plot) that corresponds to a significant increase of the value of
the measure.
*******************************************************************
* Among all indices:
* 12 proposed 2 as the best number of clusters
* 7 proposed 3 as the best number of clusters
* 2 proposed 4 as the best number of clusters
* 3 proposed 5 as the best number of clusters
***** Conclusion *****
* According to the majority rule, the best number of clusters is 2
*******************************************************************
Figure 38: Principal component analysis on the coordinates of the long aptamer doplong7 (starting without A38•T37 bp) in the unrestrained microsecond trajectory. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points. (B) Score plots along the first two principal components, colored by kmeans clusters. (C) Sum of absolute loadings of residues for the first two first principal components
Figure 39: Principal component analysis on the coordinates of the long aptamer doplong8 (starting with A38•T37 bp) in the unrestrained microsecond trajectory. Scree plot (A) showing the contribution of each principal component on the total variance and the cumulative variance labelled on selected data points. (B) Score plots along the first two principal components, colored by kmeans clusters. (C) Sum of absolute loadings of residues for the first two first principal components
pcadoplong7: The A38•T37 base pair is initially absent but canonically formed in cluster 2. However, the latter loses the C24•G29 G25•G28 base pairs. The two events are not directly linked as they occur around half a microsecond from one another.
pcadoplong8: The second cluster is characterized by the zipping of the 15:17/33:35 region (between the binding site, above, and the duplex stem, below), while the 24:25 nucleotides flip out of the loop region. In each panel, the starting cluster is characterized by the interaction between T34 (green; foreground) with G34, which is the expected base pair with restraints. Note the difference in backbone geometry.
Superimposed coordinates of the cluster centroids, aligned by their duplex stem. Clusters 1 and 2 and on the left (with blue dopamine) and right (with orange dopamine), respectively.
Initially, G9 is paired to G13, with G36 forming an orthogonal contact to the latter (left). In the second cluster’ centroid coordinates, G9 is flipped out and G36 is coplanar to G13, hence forming a more canonical base pair (right).
Figure 40
Anosova, Irina, Ewa A. Kowal, Matthew R. Dunn, John C. Chaput, Wade D. Van Horn, and Martin Egli. 2015. “The Structural Diversity of Artificial Genetic Polymers.”Nucleic Acids Research 44 (3): 1007–21. https://doi.org/10.1093/nar/gkv1472.
B. J., Grant, Rodrigues A. P. C., ElSawy K. M., McCammon J. A., and Caves L. S. D. 2006. “Bio3D: An r Package for the Comparative Analysis of Protein Structures.” 22.
Berendsen, H. J. C., J. P. M. Postma, W. F. van Gunsteren, A. DiNola, and J. R. Haak. 1984. “Molecular Dynamics with Coupling to an External Bath.”The Journal of Chemical Physics 81 (8): 3684–90. https://doi.org/10.1063/1.448118.
Case, David A., Hasan Metin Aktulga, Kellon Belfon, David S. Cerutti, G. Andrés Cisneros, Vinícius Wilian D. Cruzeiro, Negin Forouzesh, et al. 2023. “AmberTools.”Journal of Chemical Information and Modeling 63 (20): 6183–91. https://doi.org/10.1021/acs.jcim.3c01153.
Charrad, Malika, Nadia Ghazzali, Véronique Boiteau, and Azam Niknafs. 2014. “NbClust: An r Package for Determining the Relevant Number of Clusters in a Data Set” 61. https://www.jstatsoft.org/v61/i06/.
Gesteland, R. F., T. Cech, and J. F. Atkins. 2006. The RNA World: The Nature of Modern RNA Suggests a Prebiotic RNA World. Cold Spring Harbor Monograph Series. Cold Spring Harbor Laboratory Press. https://books.google.fr/books?id=3mREVdXNzFcC.
Götz, Andreas W., Mark J. Williamson, Dong Xu, Duncan Poole, Scott Le Grand, and Ross C. Walker. 2012. “Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born.”Journal of Chemical Theory and Computation 8 (5): 1542–55. https://doi.org/10.1021/ct200909j.
Izadi, Saeed, Ramu Anandakrishnan, and Alexey V. Onufriev. 2014. “Building Water Models: A Different Approach.”The Journal of Physical Chemistry Letters 5 (21): 3863–71. https://doi.org/10.1021/jz501780a.
Laskowski, Roman A., and Mark B. Swindells. 2011. “LigPlot+: Multiple LigandProtein Interaction Diagrams for Drug Discovery.”Journal of Chemical Information and Modeling 51 (10): 2778–86. https://doi.org/10.1021/ci200227u.
Le Grand, Scott, Andreas W. Götz, and Ross C. Walker. 2013. “SPFP: Speed Without CompromiseA Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations.”Computer Physics Communications 184 (2): 374–80. https://doi.org/10.1016/j.cpc.2012.09.022.
Lemieux, S. 2002. “RNA Canonical and Non-Canonical Base Pairing Types: A Recognition Method and Complete Repertoire.”Nucleic Acids Research 30 (19): 4250–63. https://doi.org/10.1093/nar/gkf540.
LEONTIS, NEOCLES B., and ERIC WESTHOF. 2001. “Geometric Nomenclature and Classification of RNA Base Pairs.”RNA 7 (4): 499–512. https://doi.org/10.1017/s1355838201002515.
Li, Zhen, Lin Frank Song, Pengfei Li, and Kenneth M. Merz. 2020. “Systematic Parametrization of Divalent Metal Ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB Water Models.”Journal of Chemical Theory and Computation 16 (7): 4429–42. https://doi.org/10.1021/acs.jctc.0c00194.
Love, Olivia, Rodrigo Galindo-Murillo, Marie Zgarbová, Jiří Šponer, Petr Jurečka, and Thomas E. Cheatham. 2023. “Assessing the Current State of Amber Force Field Modifications for DNA─2023 Edition.”Journal of Chemical Theory and Computation 19 (13): 4299–4307. https://doi.org/10.1021/acs.jctc.3c00233.
Lu, Xiang-Jun. 2020. “DSSR-Enabled Innovative Schematics of 3D Nucleic Acid Structures with PyMOL.”Nucleic Acids Research, May. https://doi.org/10.1093/nar/gkaa426.
Lu, Xiang-Jun, Harmen J. Bussemaker, and Wilma K. Olson. 2015. “DSSR: An Integrated Software Tool for Dissecting the Spatial Structure of RNA.”Nucleic Acids Research, July, gkv716. https://doi.org/10.1093/nar/gkv716.
Machado, Matías R., and Sergio Pantano. 2020. “Split the Charge Difference in Two! A Rule of Thumb for Adding Proper Amounts of Ions in MD Simulations.”Journal of Chemical Theory and Computation 16 (3): 1367–72. https://doi.org/10.1021/acs.jctc.9b00953.
R Core Team. 2023. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing. https://www.R-project.org/.
Richardson, Neal, Ian Cook, Nic Crane, Dewey Dunnington, Romain François, Jonathan Keane, Dragoș Moldovan-Grünfeld, Jeroen Ooms, Jacob Wujciak-Jens, and Apache Arrow. 2024. Arrow: Integration to ’Apache’ ’Arrow’. https://github.com/apache/arrow/.
Romanowska, Julia, Krzysztof S. Nowiński, and Joanna Trylska. 2012. “Determining Geometrically Stable Domains in Molecular Conformation Sets.”Journal of Chemical Theory and Computation 8 (8): 2588–99. https://doi.org/10.1021/ct300206j.
Salomon-Ferrer, Romelia, Andreas W. Götz, Duncan Poole, Scott Le Grand, and Ross C. Walker. 2013. “Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald.”Journal of Chemical Theory and Computation 9 (9): 3878–88. https://doi.org/10.1021/ct400314y.
Schmit, Jeremy D., Nilusha L. Kariyawasam, Vince Needham, and Paul E. Smith. 2018. “SLTCAP: A Simple Method for Calculating the Number of Ions Needed for MD Simulation.”Journal of Chemical Theory and Computation 14 (4): 1823–27. https://doi.org/10.1021/acs.jctc.7b01254.
Shen, X., B. Gu, S. A. Che, and F. S. Zhang. 2011. “Solvent Effects on the Conformation of DNA Dodecamer Segment: A Simulation Study.”The Journal of Chemical Physics 135 (3). https://doi.org/10.1063/1.3610549.
Sindhikara, Daniel J., Seonah Kim, Arthur F. Voter, and Adrian E. Roitberg. 2009. “Bad Seeds Sprout Perilous Dynamics: Stochastic Thermostat Induced Trajectory Synchronization in Biomolecules.”Journal of Chemical Theory and Computation 5 (6): 1624–31. https://doi.org/10.1021/ct800573m.
Wang, Andrew H. J., Giovanni Ughetto, Gary J. Quigley, and Alexander Rich. 1987. “Interactions Between an Anthracycline Antibiotic and DNA: Molecular Structure of Daunomycin Complexed to d(CpGpTpApCpG) at 1.2-.ANG. Resolution.”Biochemistry 26 (4): 1152–63. https://doi.org/10.1021/bi00378a025.
Wang, Junmei, Romain M. Wolf, James W. Caldwell, Peter A. Kollman, and David A. Case. 2004. “Development and Testing of a General Amber Force Field.”Journal of Computational Chemistry 25 (9): 1157–74. https://doi.org/10.1002/jcc.20035.
Wickham, Hadley, Mara Averick, Jennifer Bryan, Winston Chang, Lucy D’Agostino McGowan, Romain François, Garrett Grolemund, et al. 2019. “Welcome to the Tidyverse” 4: 1686. https://doi.org/10.21105/joss.01686.
Zgarbová, Marie, Jiří Šponer, and Petr Jurečka. 2021. “Z-DNA as a Touchstone for Additive Empirical Force Fields and a Refinement of the Alpha/Gamma DNA Torsions for AMBER.”Journal of Chemical Theory and Computation 17 (10): 6292–6301. https://doi.org/10.1021/acs.jctc.1c00697.